
Abstract
Mitochondria provide energy for various cellular activities and are involved in the regulating of several physiological and pathological processes. Mitochondria constitute a dynamic network regulated by numerous quality control mechanisms; for example, division is necessary for mitochondria to develop, and fusion dilutes toxins produced by the mitochondria. Mitophagy removes damaged mitochondria. The etiologies of peripheral neuropathy include congenital and acquired diseases, and the pathogenesis varies; however, oxidative stress caused by mitochondrial damage is the accepted pathogenesis of peripheral neuropathy. Regulation and control of mitochondrial quality might point the way toward potential treatments for peripheral neuropathy. This article will review mitochondrial quality control mechanisms, their involvement in peripheral nerve diseases, and their potential therapeutic role.
Introduction
Mitochondria generate the ATP that energizes cellular processes. In addition to oxidative phosphorylation and the tricarboxylic acid cycle, mitochondria regulate calcium homeostasis, membrane potential, production, and quarantine of reactive oxygen species (ROS), cellular metabolism, inheritance of mitochondrial DNA (mtDNA), and apoptosis (van der Bliek et al., 2017). Mitochondria maintain their networks through highly regulated quality control mechanisms (Ma et al., 2020). The concept of quality control of mitochondria includes mitochondrial dynamics and mitophagy (Ni et al., 2015). Dynamics refers to mitochondrial fusion and fission (Chan, 2020).
Peripheral neurons have a high energy demand and require stable mitochondria dynamics to distribute them in axons (Safiulina and Kaasik, 2013). Mitophagy eliminates dysfunctional mitochondria by moving them to lysosomes when mitochondria are affected by mutations in the coding genes or pathogenic factors such as chemotherapeutic agents and diabetes (Youle and Narendra, 2011; Pickles et al., 2018). After mitochondrial injury, increased membrane permeability leads to the release of a large amount of ROS. This oxidative stress is the mechanism of peripheral nerve injury and the cause of pain (Flatters, 2015). Of note, Schwann cells form myelin sheaths around peripheral nerves, and their mitochondria play vital roles in axon development and functional regulation (Ino and Iino, 2017).
For these and other reasons, the dysfunction of mitochondrial quality control is thought to participate in the development of peripheral neuropathy. Researchers have achieved impressive results regarding these mechanisms.
Mechanism of Mitochondrial Quality Control
Mitophagy
Autophagy is a process in which cellular components are encapsulated in a double-layer membrane structure called the autophagosome and are digested by lysosomes (Dikic and Elazar, 2018). Depending on the contents being phagocytized, autophagy can be nonselective or selective (Xu et al., 2021).
Mitophagy, initially proposed by John J. LeMasters, is the selective phagocytosis of damaged mitochondria (Lemasters, 2005), and is mediated by specific proteins (Zaffagnini and Martens, 2016). Mitophagy has been identified in cells under normal physiological conditions (Ding and Yin, 2012). For example, mitophagy eliminates sperm-derived mitochondria, promotes maternal inheritance of mtDNA (Youle and Narendra, 2011; Roberts et al., 2016), and eliminates the mitochondria of developing erythrocytes (Pickles et al., 2018). Peripheral neurons are postmitotic and nonproliferating cells (Roberts et al., 2016); rapid elimination of dysfunctional mitochondria is probably essential for protecting neurons against oxidative damage and degeneration.
Ubiquitin-mediated mitophagy
The classical pathway of ubiquitin-mediated mitophagy is PTEN-induced putative kinase 1 (PINK1)-Parkin-mediated mitophagy (Amadoro et al., 2014). PINK1 protein is a serine/threonine kinase (Rasool and Trempe, 2018). In a physiological state, it anchors mitochondria through its N-terminal mitochondrial targeting sequence (Durcan and Fon, 2015), and it is imported into the mitochondria through the translocase of outer membrane and translocase of inner membrane complexes (Abe et al., 2000; Lazarou et al., 2012; Ashrafi and Schwarz, 2013; Eiyama and Okamoto, 2015; Liu et al., 2018a; Rasool and Trempe, 2018).
PINK1 cleaved by mitochondrial processing peptidase (MPP) in the mitochondrial matrix and presenilin-associated rhomboid-like protein (PARL) in the inner membrane (Jin et al., 2010; Deas et al., 2011; Meissner et al., 2011; Shi et al., 2011; Greene et al., 2012; Bingol and Sheng, 2016). The fragments are eventually transferred outside the mitochondria and hydrolyzed by the proteasome (Ashrafi and Schwarz, 2013).
Parkin is an E3 ubiquitin ligase in the cytoplasm with a ubiquitin-like domain (UblD) at the N-terminal (Seirafi et al., 2015). UblD inhibits the activity of Parkin under physiological conditions (Trempe et al., 2013; Wauer and Komander, 2013; Nguyen et al., 2016) (Fig. 1A).

PINK1-Parkin-mediated mitophagy.
When damaged mitochondria lose their membrane potential, PINK1 cleavage is inhibited, leading to PINK1 accumulation on the outer membrane (Narendra et al., 2010; Durcan and Fon, 2015). After PINK1 phosphorylates Ser65 in Parkin's UblD and polyubiquitin (poly-UB) chain, Parkin E3 ubiquitin ligase activity is activated and translocates to the damaged mitochondrial outer membrane (Kim et al., 2008; Narendra et al., 2010; Kondapalli et al., 2012; Koyano et al., 2014; Shiba-Fukushima et al., 2014; Okatsu et al., 2015; Sauvé et al., 2015).
Parkin generates more poly-UB chains (typical mitochondrial ubiquitin chains include lys48 and lys63, whereas atypical ones include lys11 and lys6) on mitochondria, which are subsequently phosphorylated by PINK1 (Kulathu and Komander, 2012; Ordureau et al., 2014, 2015; Lazarou et al., 2015; Seirafi et al., 2015) (Fig. 1B).
Those that recognize the ubiquitin chain and recruit LC3 to move to damaged mitochondria are called adaptor proteins, including ubiquitin-binding protein p62 (sequestosome-1, also known as SQSTM1/p62), optineurin (OPTN), nuclear dot protein 52 (NDP52), TAX1-binding protein 1 (TAX1BP1), and neighbor of BRCA1 gene protein (NBR1) (Geisler et al., 2010; Ma et al., 2020) (Fig. 1C). Eventually, with the help of adaptor proteins, autophagosomes phagocytize mitochondria, and lysosomes further digest autophagosomes. PINK1 also directly recruits adaptor proteins and mediates mitophagy (Lazarou et al., 2015; Yamada et al., 2019).
Receptor-mediated mitophagy
Several proteins on the outer mitochondrial membrane have the LC3-interacting region (LIR) and bind directly to LC3 to induce autophagy, called receptor-induced mitophagy (Hanna et al., 2012; Zhu et al., 2013; Liu et al., 2014; Yamaguchi et al., 2016). Two types of mitophagy receptors have been identified in mammals. NIP-3-like protein X (NIX/BNIP3L) and BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3) interact with Bcl-2. The other is FUN14 domain-containing protein 1 (FUNDC1) (Liu et al., 2014; Chen et al., 2016).
The elimination of mitochondria in mature red blood cells requires the mediation of NIX/BNIP3L (Schweers et al., 2007; Sandoval et al., 2008; Zhang and Ney, 2009a; Novak et al., 2010). In hypoxia-mediated mitophagy, both types of mitophagy receptors play a role (Tracy et al., 2007; Zhang and Ney, 2009b; Liu et al., 2012). NIX/BNIP3L and BNIP3 are transcriptionally regulated by hypoxia-inducible factor (HIF) and Forkhead Box O3 (FOXO3) (Sowter et al., 2001; Chinnadurai et al., 2008). FUNDC1-mediated mitophagy activity is regulated by reversible phosphorylation at the post-translational level (Feng et al., 2013).
Specifically, under normal physiological conditions, Tyr18 of FUNDC1 is phosphorylated by Src kinase, and Ser13 is phosphorylated by casein kinase 2 (CK2) (Fig. 2A); under hypoxic stress, Tyr18 and Ser13 are simultaneously dephosphorylated and activate FUNDC1-mediated mitophagy (Chen et al., 2014; Liu et al., 2014) (Fig. 2B, C). A PGAM family member 5, mitochondrial serine/threonine protein phosphatase (PGAM5), is responsible for the dephosphorylation of FUNDC1 Ser13 site, whereas CK2 antagonizes the effect of PGAM5 and inhibits mitophagy (Chen et al., 2014; Yu et al., 2020a). Unc-51 like autophagy activating kinase 1 (ULK1) is recruited to fragmented mitochondria and phosphorylates Ser-17 of FUNDC1 to regulate mitophagy (Wu et al., 2014).
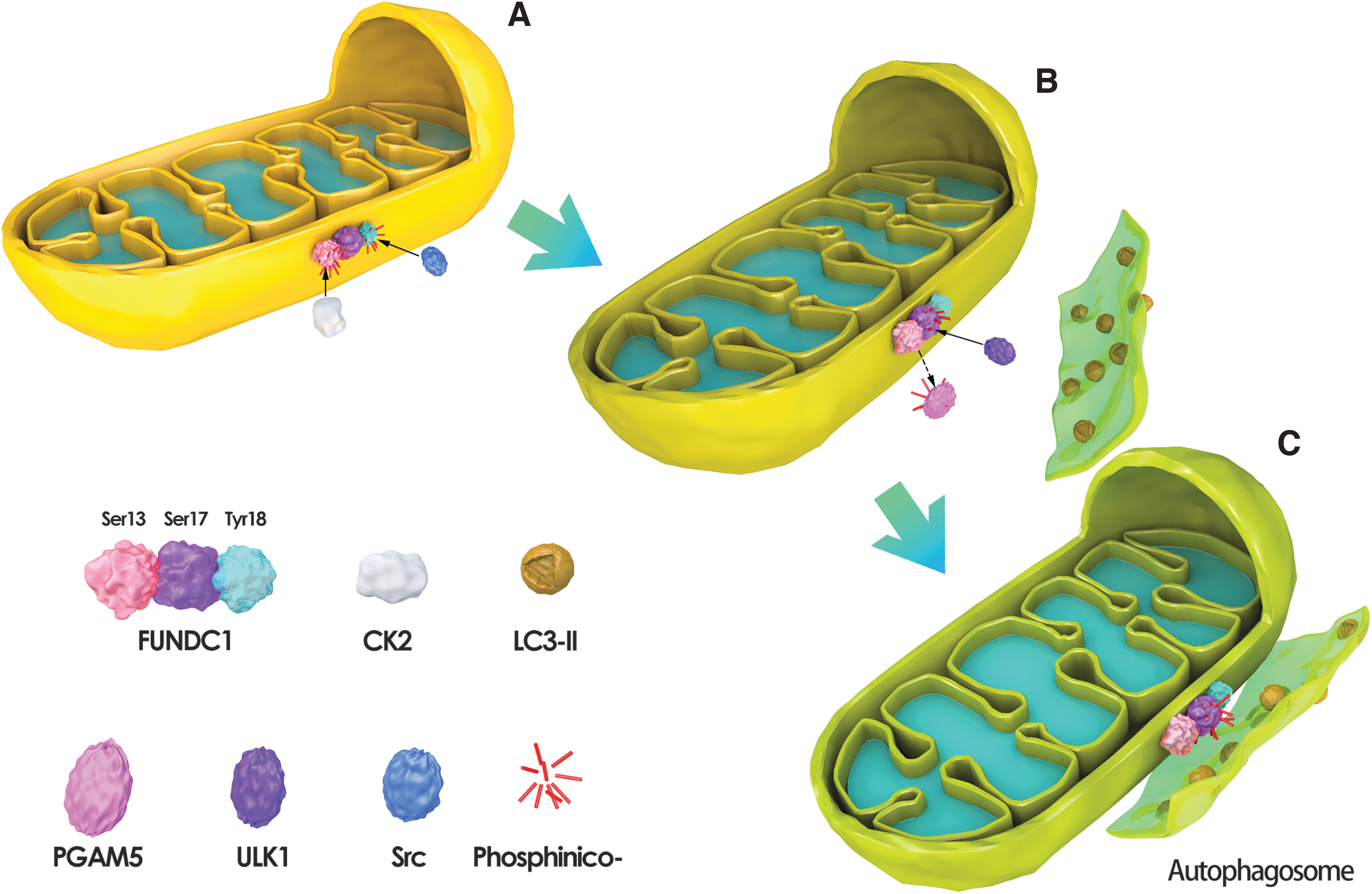
FUNDC1-mediated mitochondrial autophagy.
Mitochondrial dynamics
Under the electron microscope or fluorescence microscope, it can be dynamically observed that the mitochondrial probes disperse from one mitochondrion to two, suggesting that mitochondria are a dynamic network enterprise characterized by continuous division and fusion (Wai and Langer, 2016).
Mitochondrial dynamics maintains the morphology, number, cytoplasm, axon distribution, and physiological functions of peripheral nerve mitochondria (Meyer et al., 2017). Mitochondrial dynamics also regulate various mitochondrial functions, including mtDNA stability, apoptosis, and responses to cellular stress and mitophagy (Olichon et al., 2003; Youle and Karbowski, 2005; Twig and Shirihai, 2011; El-Hattab et al., 2018; Tong et al., 2020). In mammals, mitochondrial fusion and fission are indispensable, and even slight defects in mitochondrial dynamics can lead to disease (Chung et al., 2018).
Mitochondria fission
In mammals, an essential protein regulating mitochondrial fission is dynamic-related protein 1 (DRP1) (Mishra and Chan, 2016). Under physiological conditions, DRP1 is located in the cytoplasm (Schmitt et al., 2018). When stimulated by mitochondrial fission factors (MFFs), DRP1 is recruited to the outer membrane of mitochondria to form oligomers under the assistance of receptor proteins (Kalia et al., 2018). The oligomers form a ring structure around the mitochondria and then hydrolyze GTP with its GTPase activity to break the mitochondria apart (Wai and Langer, 2016) (Fig. 3A). The fission produces small and depolarized mitochondria that are eliminated by mitophagy (Poole et al., 2008; Twig et al., 2008; Twig and Shirihai, 2011; El-Hattab et al., 2018).
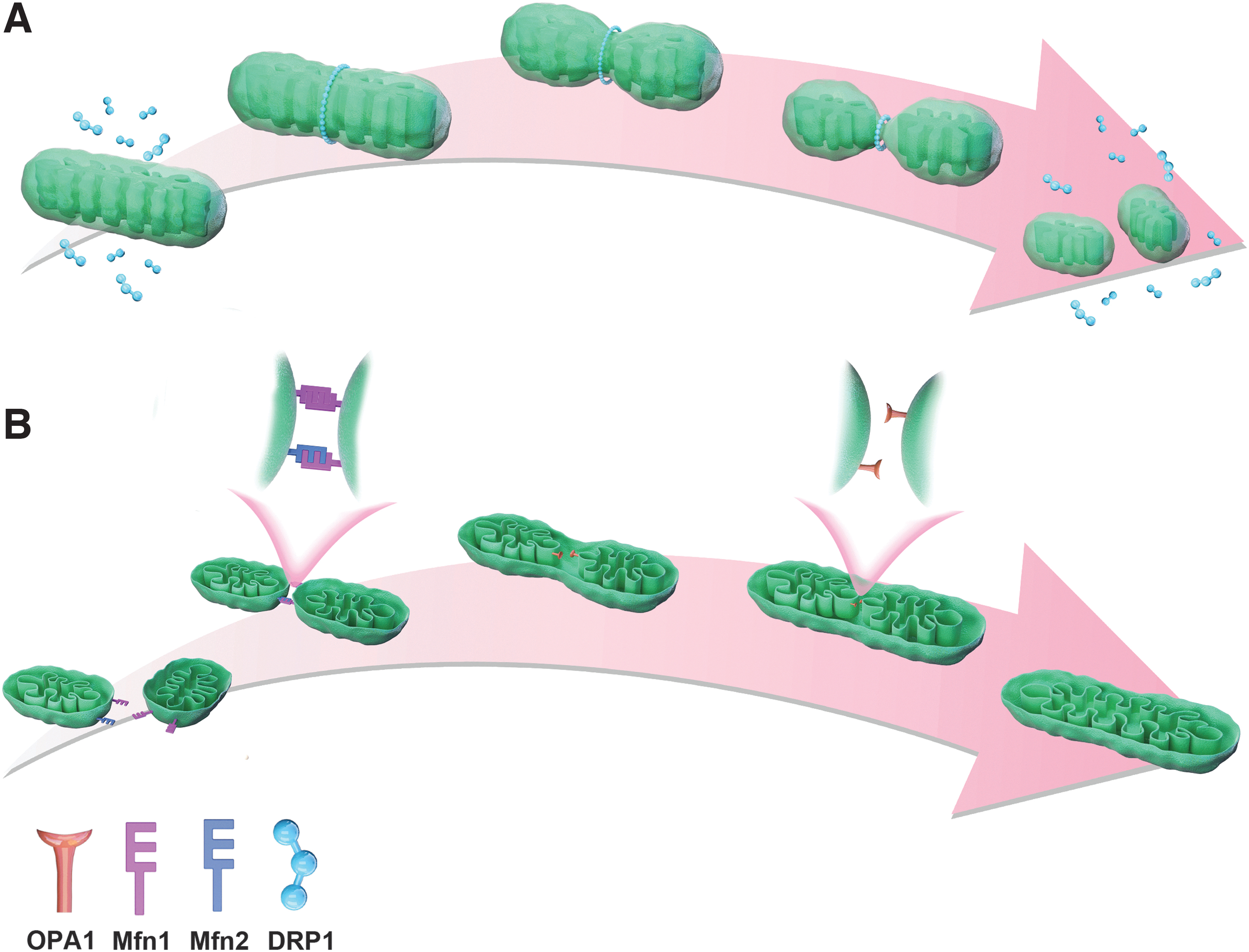
Mitochondria fission and fusion.
The receptor proteins of mitochondria include the MFF and mitochondrial dynamics 49 and 51 (MID49/MID51), whereas the role of fission protein 1 (Fis1) in mitochondrial fission of mammals is controversial (James et al., 2003; Yoon et al., 2003; Stojanovski et al., 2004; Osellame et al., 2016; Kalia et al., 2018; Yu et al., 2020b). Inhibition of DRP1 expression or phosphorylation can reduce neuronal apoptosis and protect neurons (Kim et al., 2016; Whitley et al., 2019).
Mitochondrial fusion
Because mitochondria have double-layer membranes, complete fusion requires the merging of both the outer and inner membranes. Outer membrane fusion is mediated by the mitofusins 1 and 2 (Mfn1/2) (Qi et al., 2016; Chandhok et al., 2018). First, Mfn1 and Mfn2 form a homodimer or heterodimer that promotes the formation of trans-tethering between the two adjacent outer membranes (Griffin and Chan, 2006; Qi et al., 2016). Subsequently, the two proteins exert GTPase activity to hydrolyze GTP and fuse the outer membranes of two mitochondria with conformational changes (Ryan and Stojanovski, 2012; Flippo and Strack, 2017).
Optical atrophy 1 protein (OPA1) is the core of mitochondrial inner membrane fusion, and it is involved in maintaining the shape of mitochondrial cristae (Olichon et al., 2002; Cogliati et al., 2013; Patten et al., 2014). Fusion of the inner membrane is induced when OPA1 is present on any of the trans-tethered mitochondria (Alexander et al., 2000; Cipolat et al., 2004; Meeusen et al., 2004) (Fig. 3B). Although the mechanism by which OPA1 induces inner membrane formation has not been fully established, a study reported that OPA1 needs to be hydrolyzed by a protease to form a short isoform before it can exert its action (Wang et al., 2021).
Molecular mechanisms upstream of mitochondrial quality regulation
The regulation of mitochondrial quality in peripheral nerves is not an isolated molecular mechanism, and it is often combined with other organelles or molecules in the cell to maintain cell homeostasis. The molecular mechanisms upstream of mitochondrial quality regulation include oxidative stress, endoplasmic reticulum stress, unfolded proteins, Ca2+, and inflammation. However, mitochondrial quality regulation is the most critical link, which can determine the fate of peripheral nerve cells. Refer Table 1 for related molecular pathways and specific mechanisms.
Upstream Incentives and Mechanisms of Mitochondrial Quality Regulation
BNIP3, BCL2/adenovirus E1B 19 kDa interacting protein 3; DRP1, dynamic-related protein 1; ERS, endoplasmic reticulum stress; FOXO3, Forkhead Box O3; HIF, hypoxia-inducible factor; Mfn1, mitofusins 1; Mfn2, mitofusins 2; mtDNA, mitochondrial DNA; NIX/BNIP3L, NIP-3-like protein X; OPA1, optical atrophy 1 protein; p62/SQSTM1, sequestosome-1; PINK1, PTEN-induced putative kinase 1; ROS, reactive oxygen species.
Mitochondrial Quality Control and Diseases
Congenital peripheral neuropathy
Eighteen mitochondrial proteins are encoded by mtDNA, and the nuclear genome encodes the remainder (Nissanka and Moraes, 2018). Most of these proteins are distributed in the bilayer membrane of mitochondria and regulate the mitochondrial network's regular operation together with cytoplasmic proteins transferred to mitochondria (Baloh, 2008).
Mutations of mitochondrial coding genes affect the formation and development of mitochondria and affect the transport, distribution, and function of mitochondria. These mechanisms mediate mitochondrial diseases (a review has been discussed) (Whitley et al., 2019) and congenital peripheral neuropathy.
The most typical disease is gastrocnemius atrophy (Charcot–Marie–Tooth disease type 2A, or CMT2A), caused by the mutation of the Mfn2 gene regulating mitochondrial fusion (Dorn, 2020). Patients may have motor and sensory abnormalities of peripheral nerves such as muscle weakness and atrophy of the lower limbs, mild loss of sensation, or absence of deep tendon reflex; nerve conduction velocities are normal or near normal (Lawson et al., 2005). The pathology of the disease is complex, genetic heterogeneity is high, and the specific pathogenesis is not fully understood.
Studies have shown that the 94 amino acid residue upstream of the GTPase domain of Mfn2 is most prone to mutation, and codons are mutated to R94W and R94Q (Franco et al., 2016). Thus, mitochondrial fusion is inhibited, causing its shape to become spherical or elliptical. These shapes do not readily translocate in axons, affecting the energy production of the axon and the function of synapses and eventually leading to axonal degeneration.
Ueda and Ishihara, (2018) found that Marf (Marf is the Drosophila homolog of Mfn2) mutants in the GTPase domain or mutants in the HB1 region in CMT2A Drosophila model had larger mitochondria in neurons. The authors suggested that some pathogenic mutations of Mfn2 could enhance mitochondrial fusion, indicating that the enhancement of mitochondrial fusion could also lead to pathological changes in CMT2A. Another study found that many hyper-tubular mitochondria in the primary fibroblasts of patients with hereditary spastic paraplegia hindered the transport of mitochondria to the axonal end (Lavie et al., 2017). The reason is that mutations of receptor expression-enhancing protein 1 (REEP1), which encodes tubulin, impair interactions between REEP1 and PGAM5, leading to hyperphosphorylation of mitochondrial fission protein DRP1.
Epigenetic disorders of the proteins that regulate fusion and fission also cause congenital peripheral neuropathy, providing novel insights into the disease's molecular mechanisms and therapeutic targets. Other diseases caused by mitochondrial gene mutations are characterized by central nervous system damage accompanied by peripheral neuropathy (Gao et al., 2017); it is clear that once the balance of mitochondrial fusion and fission is broken, there follows a series of pathological reactions.
Peripheral neuropathy caused by chemotherapeutic agents
Chemotherapy-induced peripheral neuropathy (CIPN) is a profound dose-dependent toxic side effect of many anti-cancer medications. These chemotherapeutic agents include platinum compounds, taxanes, and vinca alkaloids (Jaggi and Singh, 2012; Carozzi et al., 2015). The most common clinical symptoms are glove-and-sock-like paresthesias (Pulvers and Marx, 2017). In more severe cases, neuropathic pain may occur (Kerckhove et al., 2017). The mechanisms of CIPN include changes in ion channels, increased apoptosis, and the release of inflammatory cytokines (Starobova and Vetter, 2017).
Chemotherapeutic agents have a tendency to accumulate in the dorsal root ganglion (DRG) and form adducts with mtDNA, leading to mitochondrial damage and the release of a large amount of ROS (Trecarichi and Flatters, 2019). Therefore, mitochondrial dysfunction and oxidative stress are other prominent causes of CIPN and peripheral nerve pain (Waseem et al., 2018).
Some central degenerative diseases, such as Parkinson's disease, the pathogenesis is mitochondrial dysfunction caused by PINK1 and Parkin mutations leading to the loss of substantia nigra dopaminergic neurons (Dar et al., 2020; Malpartida et al., 2021). As for CIPN, people have also found that mitochondrial damage and oxidative stress can cause neuropathy (Lim et al., 2015; Rumora et al., 2019; Trecarichi and Flatters, 2019). Therefore, investigators further conjectured the relationships between mitochondrial quality control and CIPN and experimented to obtain meaningful results.
Kober et al. (2018) measured the expression of mitochondrial genes involved in peripheral neuropathy in the peripheral blood of breast cancer survivors receiving paclitaxel chemotherapy; the authors found that mitochondrial fission-related genes were linked to peripheral nerve pain. The same phenomenon was also noted in primary mouse DRG cells treated with vincristine, with reduced mitochondrial fusion and, to a lesser extent, mitophagy (Chen et al., 2020).
In another study, Drosophila larvae were treated with paclitaxel to induce peripheral neuropathy. Overexpression of PINK1 in class IV dendritic branched sensory neurons improved thermal hyperalgesia of the model, whereas PINK1 knockout led to an increase in thermal hyperalgesia (Kim et al., 2020). Nevertheless, the molecular mechanisms of these phenomena have not been elucidated.
After knocking down parkin in PC12 (a rat pheochromocytoma cell line) and treating with cisplatin, Zhang et al. (2019) found that the toxicity of cisplatin to PC12 increased, which was primarily owing to the damage of PINK1/parkin mitochondrial autophagy pathway, resulting in mitochondrial dysfunction, ATP deficiency, and ROS burst, eventually increasing apoptosis; overexpression of parkin enhanced mitochondrial autophagy and reversed the damage. Song and colleagues used tricresol phosphate (TOCP) to treat Neuro-2a (N2a, a mouse neuroblastoma cell line) to mimic organophosphate-induced delayed neuropathy, showing that TOCP activated the PINK1/parkin-mediated mitochondrial autophagy pathway in N2a cells, and the protein p62 was identified as the mitochondrial autophagy adapter (Wang et al., 2019).
The inhibition of mitochondrial autophagy in gastrocnemius muscle after sciatic nerve transection in mice improved symptoms of paralysis in that muscle (Graham et al., 2018), suggesting that mitochondrial dysfunction may be the common pathogenic mechanism of peripheral neuropathy caused by chemotherapeutic agents and other toxic drugs. Whether mitophagy mediated by PINK1/Parkin plays a protective role may be related to the type of histopathology and severity. More preclinical trials are needed to determine whether the damaging medications activate ubiquitin-mediated mitophagy, whether the adapters are the same, and through what mitochondrial mechanism autophagy alleviates chemotherapy-induced peripheral neuropathic pain.
Diabetic peripheral neuropathy
Diabetic polyneuropathy (DPN) is the most common chronic complication of diabetes mellitus (Feldman et al., 2017). The underlying mechanisms of DPN have various pathways, including the polyol pathway, the deposition of advanced glycosylation products, the hexosamine pathway, and the protein kinase C pathway (Ceriello, 2003; Sifuentes-Franco et al., 2017). The final common endpoint is that oxidative stress, nitrosation stress, and the reduction of neuronal antioxidant defense mechanisms lead to diseases (Bandeira et al., 2013; Stavniichuk et al., 2014). The signs and symptoms of DPN include decreased blood flow to supplying nerves, anoxia of the endoneurium, degeneration of motor nerves, loss of sensation, and neuropathic pain (Llewelyn, 2003; Peltier et al., 2014; Wang et al., 2014).
Several studies noted that mitochondrial quality control participates in DPN, and regulating the expression of mitochondrial quality control-related proteins effectively inhibits DPN development and alleviates the peripheral nerve pain caused by diabetes.
Yoon and colleagues found that the morphology of mitochondria was tied to ROS content after high-glucose treatment, and inhibition of mitochondrial division reduced ROS production (Yu et al., 2006). The same conclusion was reached in a subsequent experiment in which primary rat DRG cells were cultured with high-glucose in vitro; it was also demonstrated that high glucose increased the expression of mitochondrial fission protein DRP1 and the colocalization of DRP1 and Bax, suggesting that the interaction between mitochondrial fission and apoptosis leads to DPN (Leinninger et al., 2006). However, the apoptosis pathway of DRP1/Bax is different from that of BIM/Bax (Leinninger et al., 2006).
In a recent study, in a mouse model of type I diabetes, the upregulation of p53 in the ruined DRG inhibited the transcription of Parkin (Yamashita et al., 2019). The subsequent disruption of Parkin-mediated mitochondrial quality control mechanism led to ROS accumulation and ultimately to DPN and diabetic neuropathic pain (Yamashita et al., 2019). The process could be reversed using p53 selective inhibitors, consistent with the negative regulation of p53 on Parkin in cultured cortical neurons in vitro (da Costa et al., 2009).
Although investigators have long recognized that mitochondrial dysfunction is the pathogenic mechanism of DPN, the similarities and differences between mitochondrial quality control in types 1 and 2 diabetes have not yet been conclusively established, and targeted medications for mitochondrial quality control need to be further explored.
Treatment
At present, treatment of peripheral neuropathy focuses on the cure of primary disease or alleviating symptoms; there is no definitive treatment. With the in-depth exploration of the mitochondrial quality control mechanism and its relationship with peripheral neuropathy, some new therapeutic agents and methods have been explored. Some medications that have been used clinically for other indications can be used to treat peripheral neuropathy by regulating mitochondrial quality control. Because each disease causes different mitochondrial quality control disorders, the treatment methods are different. According to existing preclinical experiments, treatment methods include gene therapy, drug therapy (peptides, agonists/inhibitors, mitochondrially targeted drugs, antioxidants), and physical therapy (Table 2).
List of Methods to Treat Peripheral Neuropathy by Regulating Mitochondrial Quality
AAV, adeno-associated virus; CCI, chronic constriction injury; CIPN, chemotherapy-induced peripheral neuropathy; CMT2A, Charcot–Marie–Tooth disease type 2A; DPN, diabetic polyneuropathy; Fis1, fission protein 1; HBOT, hyperbaric oxygen therapy; HDAC6, histone deacetylase 6; IGF-1, insulin-like growth factor 1; MEFs, murine embryonic fibroblasts; MitoQ, mitoquinone; MT, mito-TEMPO; N2a, neuro-2a; Prp, prion protein; STZ, streptozotocin.
Gene therapy
Some cases of congenital peripheral neuropathy are caused by mutations in genes encoding mitochondrial proteins. Therefore, gene therapy for mutated genes is a fundamental treatment. SLC25A46 is a mitochondrial protein encoded by the nuclear genome; its deletion leads to mitochondrial hyperfusion and causes ataxia. Huang et al. packaged Slc25a46 with adeno-associated virus (AAV) and injected it into Slc25a46−/− mice circulation (Yang et al., 2020). As a result, mitochondrial dynamics were restored, and the function of the respiratory chain improved.
Peripheral neuropathy caused by mutations in a single mtDNA or nucleus-encoded mitochondrial gene can be considered for gene-targeted therapy. AAV-mediated gene therapy in vivo has high transduction efficiency (Colella et al., 2018), and successfully applied in degenerative neuropathy (Piguet et al., 2017; Perez et al., 2020). However, there are few studies on AAV-mediated gene therapy of congenital peripheral neuropathy in vivo, and its effective gene targets and immunogenicity are even more challenging (Verdera et al., 2020).
Baloh and colleagues used transgenic technology to express Mfn1 in the nervous system of Mfn2R94Q mice, which not only rescued the stunted growth but also increased their survival rate (Zhou et al., 2019). Their experiments also proved that the Mfn1/Mfn2 ratio is a crucial determinant of the tissue specificity of CMT2A, and Mfn1 is the treatment target (Zhou et al., 2019). This provides a theoretical basis for the application of gene therapy CMT2A.
Drug therapy
Peptides
Scientists have invented some peptides that can enter the cell to regulate the interaction between molecules and then change the protein conformation to achieve treatment (Barbullushi et al., 2019). Rocha et al. (2018) used HR1 minipeptides to make Mfn2 a definitive transition to the fusion-permissive conformation (Franco et al., 2016). and reversed the mitochondrial axon transport disorder in the sciatic nerve of CMT2A mice. For streptozotocin (STZ)-induced type 1 diabetic rats, Paul Aghanoori believes that insulin-like growth factor 1 (IGF-1) can restore mitochondrial dysfunction and relieve peripheral nerve pain (Aghanoori et al., 2019). But there are no experiments to point out the difference between mitochondrial quality control disorders in type 1 and type 2 diabetes, and there are no specific drugs to alleviate DPN.
Agonists/inhibitors
Recently, Mfn2 and histone deacetylase 6 (HDAC6) have become research hotspots in the pathogenesis of CMT2A. Gerald W. Dorn II and others successfully used Mfn2 agonist (also a mitomycin receptor agonist) to promote the trans-Mfn-Mfn dimer formation and promote mitochondrial fusion in vivo and in vitro (Rocha et al., 2018; Dang et al., 2020). Another study used HDAC6 inhibitor (SW-100) to restore α-tubulin acetylation and improved mice's motor and sensory dysfunction (Picci et al., 2020). It puts forward that HDAC6 is a target to ameliorate the CMT2A phenotype and provides new ideas for applying epigenetics in the treatment of CMT2A.
Mitochondrial-targeted drugs and nonmitochondrial-targeted antioxidants
The efficacy of mito-TEMPO, a mitochondrial-targeted drug, was demonstrated in a variety of pathological types (Liu et al., 2018b; Shetty et al., 2019). It inhibits oxidative stress by increasing the content of reduced glutathione and enhancing total superoxide dismutase activity. One study found that mito-TEMPO enhanced the expression of mitochondrial fusion genes and inhibited the expression of fission genes, and alleviated peripheral neuralgia in a rat model of chronic constriction injury (CCI) (Zhan et al., 2018).
Mitoquinone (MitoQ), an anti-neoplastic agent, enhanced mitochondrial fusion genes' expression, inhibited fission genes' expression, and alleviated vincristine-induced pain in mice (Chen et al., 2020). Melatonin, tanshinone IIA, salidroside, and other nonmitochondrial-targeted antioxidants enhanced ubiquitin-mediated mitochondrial autophagy (Areti et al., 2017; Li and Chen, 2019; Gu et al., 2020). They alleviated CIPN pain without causing deterioration of the original tumor in vivo and in vitro studies (Areti et al., 2017). But nowadays, there are few reports about antioxidants regulating mitochondrial dynamics to treat peripheral neuropathy, and no medication combinations have been reported to regulate mitochondrial quality control.
Physical therapy
Hyperbaric oxygen therapy (HBOT) is 100% oxygen breathing in a pressure vessel above atmospheric pressure, a well-established treatment for various conditions (Memar et al., 2019). HBOT has been proven effective in treating carbon monoxide poisoning, crush injuries, and infections. It has also been reported that HBOT can reduce neuromuscular damage (Moghadam et al., 2020). Some people have observed that HBOT can regulate mitochondrial autophagy to reduce pain in CCI rats (Han et al., 2017; Kun et al., 2019), but how HBOT can regulate peripheral nerve mitochondrial quality needs to be further explored.
Conclusions and Outlook
Peripheral neuropathy is related to the patient's quality of life and psychology (Seretny et al., 2014), so it has always been the medical community's focus (Barrell and Smith, 2019). Both mitophagy and mitochondrial dynamics are indispensable in maintaining the normal physiological functions of peripheral nerves, and they are also an essential link in the pathogenesis of peripheral neuropathy.
The relationship between mitochondrial dysfunction and peripheral neuropathy has been identified. Beneficial mitochondrial quality regulation can eliminate dysfunctional mitochondria to achieve the purpose of treatment, but there is no uniform and specific medicine or method for the treatment of peripheral neuropathy (Derksen et al., 2017). For congenital peripheral neuropathy, the better solution would be to couple gene therapy and peptides, meaning that the treatment should address both replacing the gene and stabilizing gene expression product conformation (Barbullushi et al., 2019). For noncongenital peripheral neuropathy, such as CIPN or DPN, mitochondrial-targeted drugs and nonmitochondrial-targeted antioxidants can be combined, or maybe medication and physical therapy can be combined.
In short, more preclinical experiments are needed to prove the feasibility and safety of the combination therapy. The mechanism of mitochondrial quality regulation remains to be explored, and it is expected to become an essential target for the treatment of peripheral neuropathy.
Footnotes
Authors' Contributions
T.Z. participated in original draft and figure preparation. J.L. participated in the structured outline preparation and the article revising. T.Z. participated in the article revising. G.Z. participated in the conception of the review and the final revision of the article.
Acknowledgment
We thank Robert P. Lindeman, MD, PhD, for editing and proofreading this review.
Disclosure Statement
The authors have no potential conflicts of interest to disclose.
Funding Information
This work was supported by the National Natural Science Foundation of China (Grant No. 81903273, to G.Z.). The funding sources had no role in study conception and design, data analysis, or interpretation, article writing or deciding to submit this article for publication.