
Abstract
Diabetic kidney disease (DKD) is the leading cause of end-stage renal disease, but the molecular mechanisms of disease remain not very clear and there is no curative therapeutic strategy so far. This study was carried out to identify the expression profile of circular RNA (circRNA) in human DKD and explore circRNA regulatory function in glomeruli and tubuli simultaneously. As a result, a total of 40 upregulated and 23 downregulated differentially expressed circRNAs (DEcircRNAs) were detected. Six candidate DEcircRNAs were verified by quantitative real-time polymerase chain reaction in high glucose-treated human mesangial cells and human proximal renal tubular epithelial cells, respectively. Gene ontology and Kyoto Encyclopedia of Genes and Genomes pathway analysis revealed that both in glomeruli and in tubuli the DEcircRNAs-targeted genes participated in many pathophysiological processes of DKD. Correlation analysis with renal function showed that expression level of DEcircRNA-targeted hub gene was related to renal function. In conclusion, this is the first study to report expression profiles of circRNAs in kidney of DKD patients, and further analysis demonstrated that circRNA probably played a significant regulatory role, providing help for understanding the pathogenesis of DKD and investigating novel diagnostic and therapeutic strategy.
Introduction
Diabetic kidney disease (DKD), a major complication of diabetes mellitus, is the leading cause of end-stage renal disease and has become a public issue that seriously endangers human health (Kainz et al., 2015; Saeedi et al., 2019). The pathogenesis of DKD is complex and involves a variety of mechanisms, including genetic factors, metabolic disorder, hemodynamic changes, inflammatory reactions, oxidative stress, and so on (Lytvyn et al., 2020). Several signal transduction and biochemical mechanisms leading to DKD have been studied over the years (Kato and Natarajan, 2014; Gnudi et al., 2016), but there is no curative therapeutic strategy so far. Therefore, it is urgent to explore the underlying mechanism and develop specific and effective therapies.
Circular RNAs (circRNAs) are single-stranded covalently closed RNA molecules that are produced from pre-mRNAs through a process called back splicing (Kristensen et al., 2019). It is now recognized that circRNAs exert important biological functions, and they have been implicated in several human diseases, such as cancer, diabetes mellitus, neurological disorders, cardiovascular diseases, and so on (Hanan et al., 2017; Fang et al., 2018; Kristensen et al., 2018; Aufiero et al., 2019). With in-depth study of circRNA, some reports showed that it also played an important role in the development of DKD. A recent study showed that circRNA_010383 was validated to be downregulated in db/db mice, mouse mesangial cells (SV40-MES13), and mouse tubular epithelial cells treated with high glucose (HG, 30mM). Further investigation found that circRNA_010383 could act as a sponge for miRNA-135a, negatively regulating the expression of the transient receptor potential cation channel, subfamily C, member 1 (TRPC1), thereby promoting the synthesis of extracellular matrix (ECM)-related protein and renal fibrosis (Peng et al., 2021). In addition, Hu et al. (2019) reported that circRNA_15698 was significantly upregulated in both db/db mice and HG (25 mM)-treated mouse mesangial cells (SV40-MES13), thereby aggravating the accumulation of ECM through miR-185/TGF-β1 axis. By RNA sequencing of peripheral blood, Fang et al. (2018) demonstrated that circANKRD36 was upregulated in peripheral blood leukocytes and correlated with chronic inflammation in type 2 diabetes (T2DM), suggesting that circANKRD36 may participate in the development of DKD.
Though some studies about role of circRNA in DKD have been performed, it is worth noting that the candidate circRNAs of current studies were all identified from DKD animal, HG-treated cells, or human peripheral blood, but the landscape of circRNA in kidney of DKD patients has not been reported and regulatory role of circRNAs in both glomeruli and tubuli remained unknown to date. In this study, we first revealed circRNA expression profile in kidney of DKD patients, and further explored potential regulatory mechanism of circRNA in DKD glomeruli and tubuli simultaneously, providing help for DKD diagnosis and therapy.
Material and Methods
Ethics statement
This study was approved by the ethics committee of Tong Ren Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, PR China (Ethical Approval No. 2017-13). All studies related with humans were performed with consent from the patients.
Renal tissue collection and histopathological evaluation
All the patients were recruited from Tong Ren Hospital, Shanghai Jiao Tong University School of Medicine, and each participant underwent written informed consent. Renal cortexes of the DKD group were obtained from DKD patients diagnosed by clinical characteristics and pathological changes of renal biopsy (Tervaert et al., 2010; Stevens et al., 2013), whereas the nondiseased renal cortexes from renal carcinoma patients undergoing nephrectomy were taken as controls. After renal biopsy or surgery, parts of renal tissues were collected for histopathological evaluation, including hematoxylin–eosin (HE), periodic acid-Schiff (PAS), Masson staining, and so on. Other parts of tissues were fixed in 3% glutaraldehyde and next for electron microscopy. Remaining samples were snap frozen in liquid nitrogen as soon as possible after renal biopsy or surgery and stored at −80°C. According to pathological results, three frozen DKD tissue samples and three controls were selected for circRNA microarray assay.
RNA isolated and circRNA microarray assay
Total RNA was isolated from renal cortex tissue using the TRIzol reagent (Invitrogen, Carlsbad, CA). Then, the RNA samples were quantified using the NanoDrop 8000 (Thermo Scientific), and the RNA integrity was assessed using Agilent Bioanalyzer 2100 (Agilent Technologies).
circRNAs were assayed with the Affymetrix Clariom D human array, which contained 12,200 circRNA-specific probes. The resulting RNA samples labeling, microarray hybridization, and washing were performed based on the manufacturer's standard protocols. In brief, total RNA was transcribed to double-strand cDNA, sense strand cDNA was fragmented and biotinylated, then biotinylated cDNA was hybridized onto the microarray for 20 h in GeneChip 645 hybridization oven at 45°C. Next, the hybridized chips were washed by GeneChip Fluidics Station 450, and then the arrays were scanned by the GeneChip Scanner 3000 7G (Affymetrix).
Data files were generated and processed with Affymetrix software and expression console. Differentially expressed circRNAs (DEcircRNAs) were identified through fold change (FC) as well as p-value was calculated using t-test. Threshold set for up- and downregulated circRNAs was |FC| > = 1.5 and p-value <0.05.
Cell culture and treatment
Human mesangial cells (HMCs) and human proximal renal tubular epithelial cells (HK2) were purchased from the Cell Bank of Type Culture Collection (Chinese Academy of Sciences, Shanghai, China). HMCs were maintained in DMEM (Gibco, BRL) and HK2 in DMEM/Ham's F12 (Gibco, BRL), supplemented with 10% fetal bovine serum (Gibco, BRL), 100 U/mL penicillin (Amresco), and 100 U/mL streptomycin (Amresco), and incubated at 37°C in a humidified incubator with 5% CO2. Then 60–70% confluent cells were rendered quiescent by incubation for 14 h in serum-free medium, then were maintained in a medium containing 5.5 mM
Quantitative real-time polymerase chain reaction
Total RNA from cells was isolated using TRIzol agent (Invitrogen) and quantified with NanoDrop 8000 (Thermo Scientific). One microgram of total RNA was applied to a reverse transcription reaction using reverse transcription kit (TAKARA, No. RR047A). SYBR Green (Vazyme, No. Q511-02) was used to analyze quantitative circRNA with the QuantStudio 3 qRT-PCR system (Thermo Fisher Scientific, CA). Primer sequences are listed in Supplementary Table S1. Relative expression levels of the circRNA in cells were normalized to internal β-actin and calculated based on the 2−ΔΔCT method.
Gene expression omnibus data download and processing
The GSE30528 and GSE30529 data sets were obtained from the Gene Expression Omnibus (GEO) database (
circRNA–miRNA–mRNA interaction network construction
CircInteractome Database (
Gene ontology and pathway enrichment analyses
Gene ontology (GO) classification and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed with the clusterProfiler package (Yu et al., 2012). p < 0.05 was considered to indicate a statistically significant difference.
Protein–protein interaction network construction and hub genes identification
The STRING database (version 11.0,
Correlation analysis with renal function
Renal function data, including estimated glomerular filtration rate (GFR) of DKD patients and urinary albumin/creatinine ratio (ACR) of diabetic animal models, were obtained from Nephroseq v5 online platform (
Statistical analysis
SPSS version 22.0 was used for data analysis. All the data are expressed as means ± SEM. The expression differences were analyzed with two-tailed unpaired Student's t-test. Correlations between the parameters were analyzed by Pearson correlation analysis. A value of p < 0.05 was considered as the statistical significance.
Results
Pathological characteristics of DKD patients and controls
Three pairs of renal samples from DKD and control patients were included in our study, and clinical data of patients are listed in Supplementary Table S2. Then histopathological evaluation of samples was performed, and typical pathological changes of HE and PAS staining are shown in Figure 1. Compared with those of control, increase of the glomeruli volume, mesangial proliferation and expansion, accumulation of ECM, and nodular glomerulosclerosis could be detected in renal tissue of the DKD group.
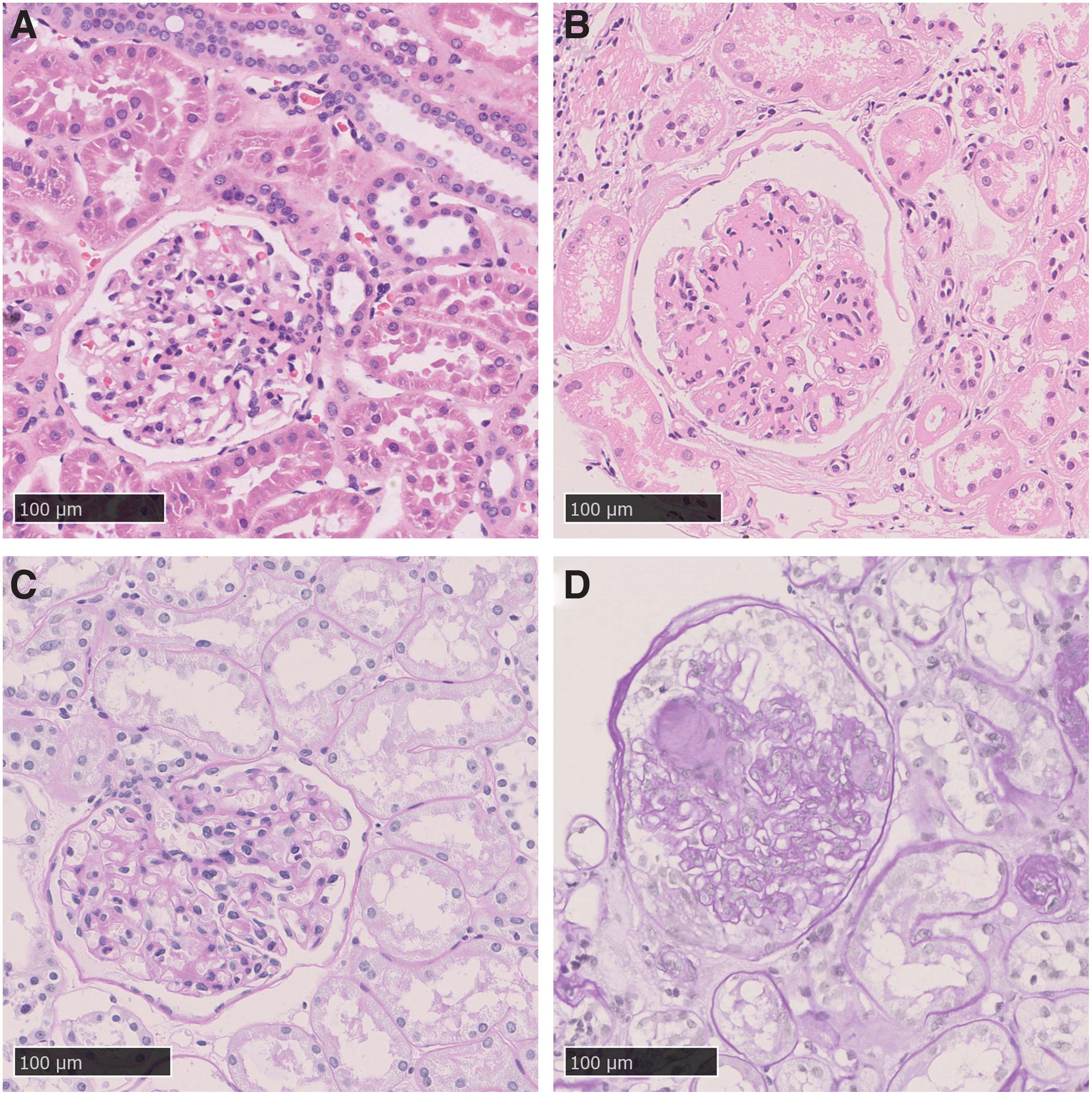
Pathological characteristics of kidney in DKD patients and controls. Representative pictures of HE staining in kidney of control
circRNA expression profiles in DKD patients and controls and analysis of DEcircRNA parental gene
Data were analyzed, normalized, and FPKM (Fragments Per Kilobase per Million mapped reads) values were visualized in boxplot (Fig. 2A), which demonstrated that data sets for the circRNA profiles of the six samples were nearly identical. A microarray analysis of 617 circRNAs was performed, there were 63 DEcircRNAs found in the DKD group compared with control according to the screening criteria (|FC| > = 1.5, p < 0.05). A heatmap of 63 DEcircRNAs was constructed to show the distinguishable circRNAs expression profiles (Fig. 2B). Among them, 40 circRNAs were upregulated and 23 circRNAs were downregulated. In addition, the distribution patterns of DEcircRNAs in the chromosomes are shown in Figure 2C, revealing that DEcircRNAs were transcribed from not only autosomes and chromosome X but also mitochondrial chromosome (chr M).

circRNA expression profiles in DKD patients and controls and DEcircRNA parental gene analysis.
Function of circRNA is closely relevant to its parental gene, therefore, GO and KEGG pathway analyses of DEcircRNA parental genes were performed. GO analyses include biological process, molecular function, and cellular component, the top 10 GO processes of each domain are shown in Figure 2D. In addition, the top 30 KEGG signaling pathways are shown in Figure 2E. The pathways not only involved focal adhesion, PI3K-Akt signaling pathway, cell cycle, and so on, but also evidently included a variety of cancer diseases.
Quantitative real-time polymerase chain reaction validation of the selected circRNAs in HG-treated HMCs and HK2
To validate the circRNA microarray data, 10 DEcircRNAs were selected and furthermore for validation by quantitative real-time polymerase chain reaction (qRT-PCR) analysis in HG-treated HMCs and HK2. HG-treated HMCs represent DKD glomerular model, whereas HK2 represent DKD tubular model. Results revealed that six circRNAs were evidently upregulated in HG-treated HMCs, two circRNAs were obviously upregulated in HG-treated HK2, and four circRNAs were distinctly downregulated in HG-treated HK2 (Fig. 3). Melt curve plots of circRNAs in qRT-PCR are shown in Supplementary Figure S1.
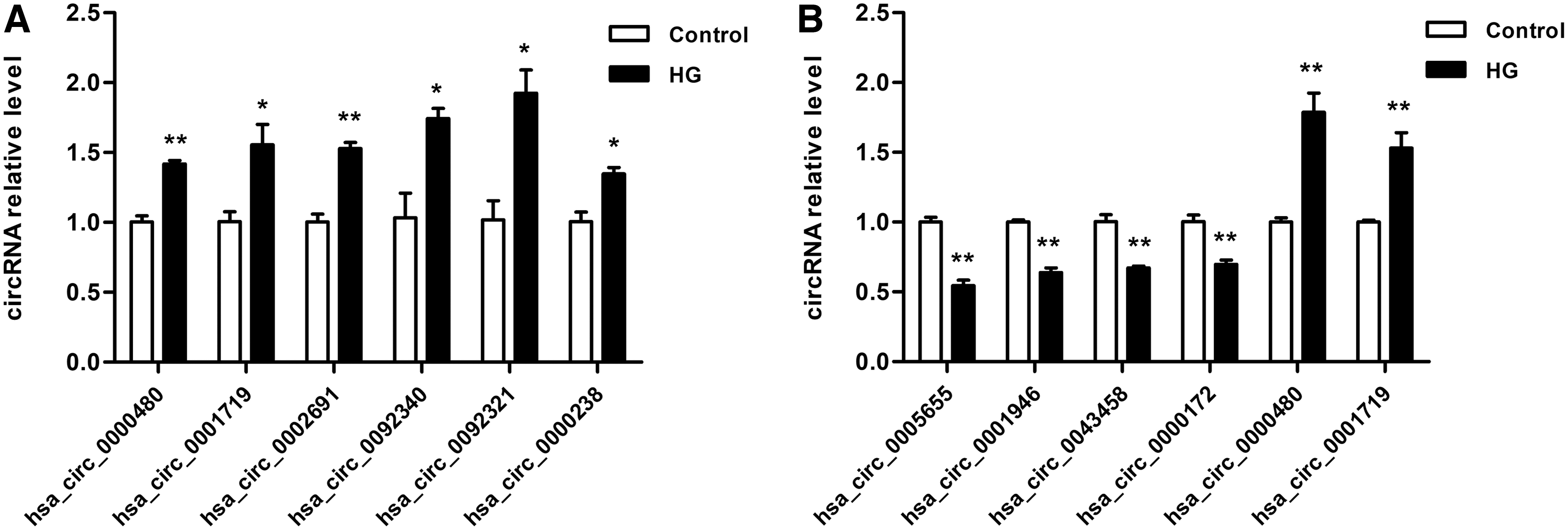
qRT-PCR validation of the selected circRNAs in HG-treated HMCs and HK2.
mRNA microarray data from glomeruli and tubuli of DKD patients and controls
Accumulating evidence has suggested that both glomerular and tubular injuries were implicated in DKD development. To explore role of circRNA in DKD glomeruli and tubuli simultaneously, GSE30528 and GSE30529 data sets were downloaded from GEO and analyzed with GEO2R tool. The box plots and volcanos of mRNA expression in glomeruli and tubuli of DKD patients and controls are presented in Figure 4. GSE30528 data set includes 9 DKD and 13 control glomerular samples, whereas GSE30529 data set includes 10 DKD and 12 control tubular samples. In total, 602 DEmRNAs were detected in glomeruli of the DKD group compared with control, including 444 upregulated and 158 downregulated mRNAs, whereas 689 DEmRNAs were detected in tubuli of the DKD group compared with control, involving 569 upregulated and 120 downregulated mRNAs.

mRNA microarray data from glomeruli and tubuli of DKD patients and controls.
CeRNA networks (DEcircRNA–DEmiRNA–DEmRNA) in DKD glomeruli and tubuli
circRNAs act through diverse mechanisms, among which serving as miRNA sponges is one of the most important ways and has been widely studied. Therefore, the ceRNA networks in glomeruli and tubuli were constructed simultaneously and shown in Figure 5. The glomerular ceRNA network contained 579 circRNA–miRNA pairs and 384 miRNA–mRNA pairs, including 57 circRNAs, 157 miRNAs, and 205 mRNAs (Fig. 5A). Meanwhile, tubular ceRNA network contained 586 circRNA–miRNA pairs and 903 miRNA–mRNA pairs, including 53 circRNAs, 149 miRNAs, and 203 mRNAs (Fig. 5B).
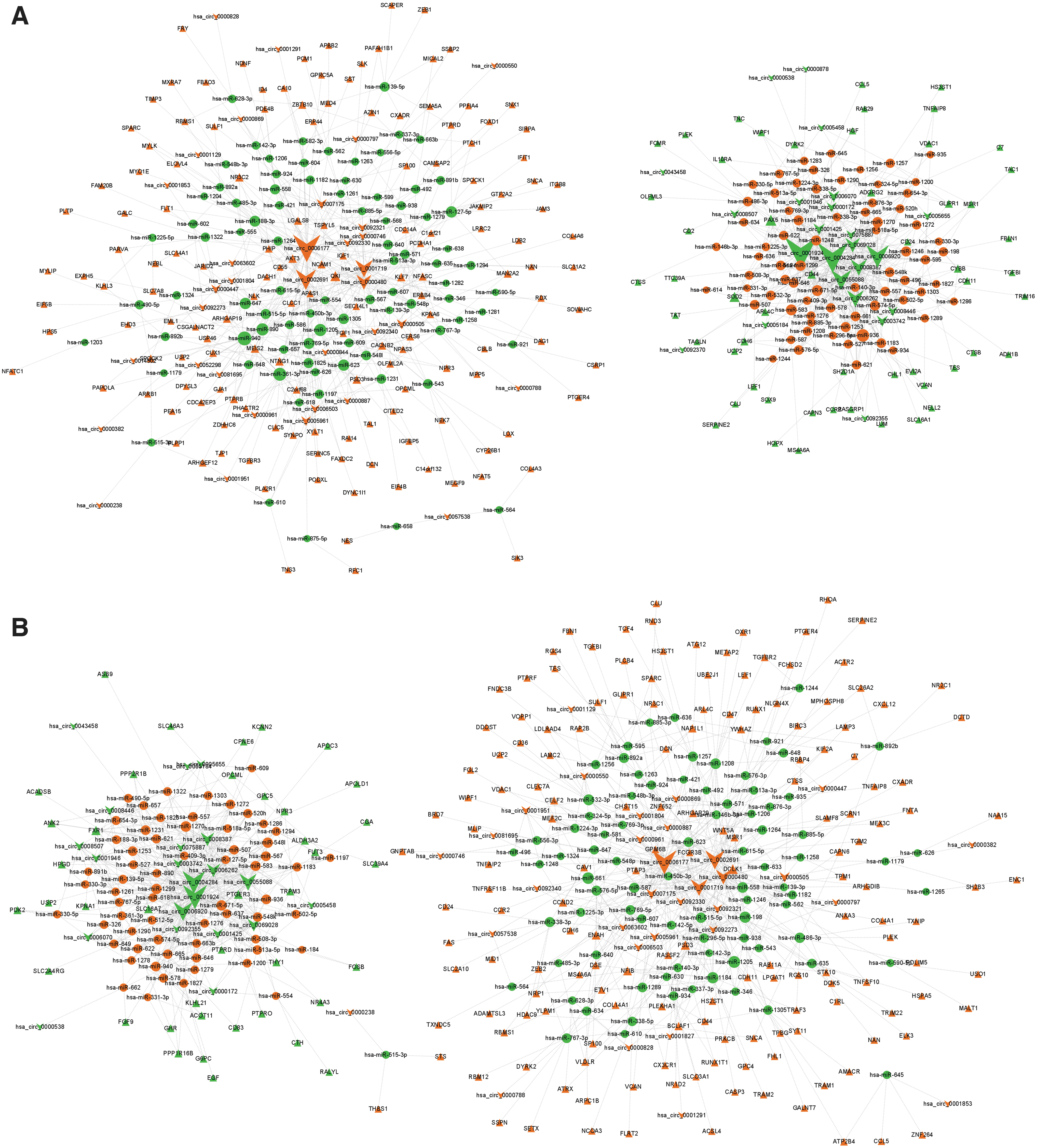
DEcircRNA–DEmiRNA–DEmRNA networks in DKD glomeruli and tubuli.
Prediction of circRNA functions through KEGG and GO analyses
To investigate the role of circRNA in glomeruli and tubuli, GO enrichment and KEGG pathway analyses were performed for genes in ceRNA networks. GO enrichment analyses of circRNA-targeted genes in glomeruli and tubuli are shown in Figure 6A and B, respectively. GO enrichment revealed that glomerular genes mostly enriched in ECM organization, collagen type IV trimer, and glycosaminoglycan binding, whereas tubular genes significantly enriched in actin cytoskeleton reorganization, neuron projection, and heparin binding. KEGG pathway analysis demonstrated that both glomerular and tubular genes obviously enriched in ECM–receptor interaction, focal adhesion, and proteoglycans in cancer. In addition, glomerular genes mostly enriched in MAPK and PI3K-Akt signaling pathway, whereas tubular genes evidently enriched in NOD-like receptor signaling pathway, Wnt signaling pathway, TGF-β signaling pathway, and NF-kappa B signaling pathway.
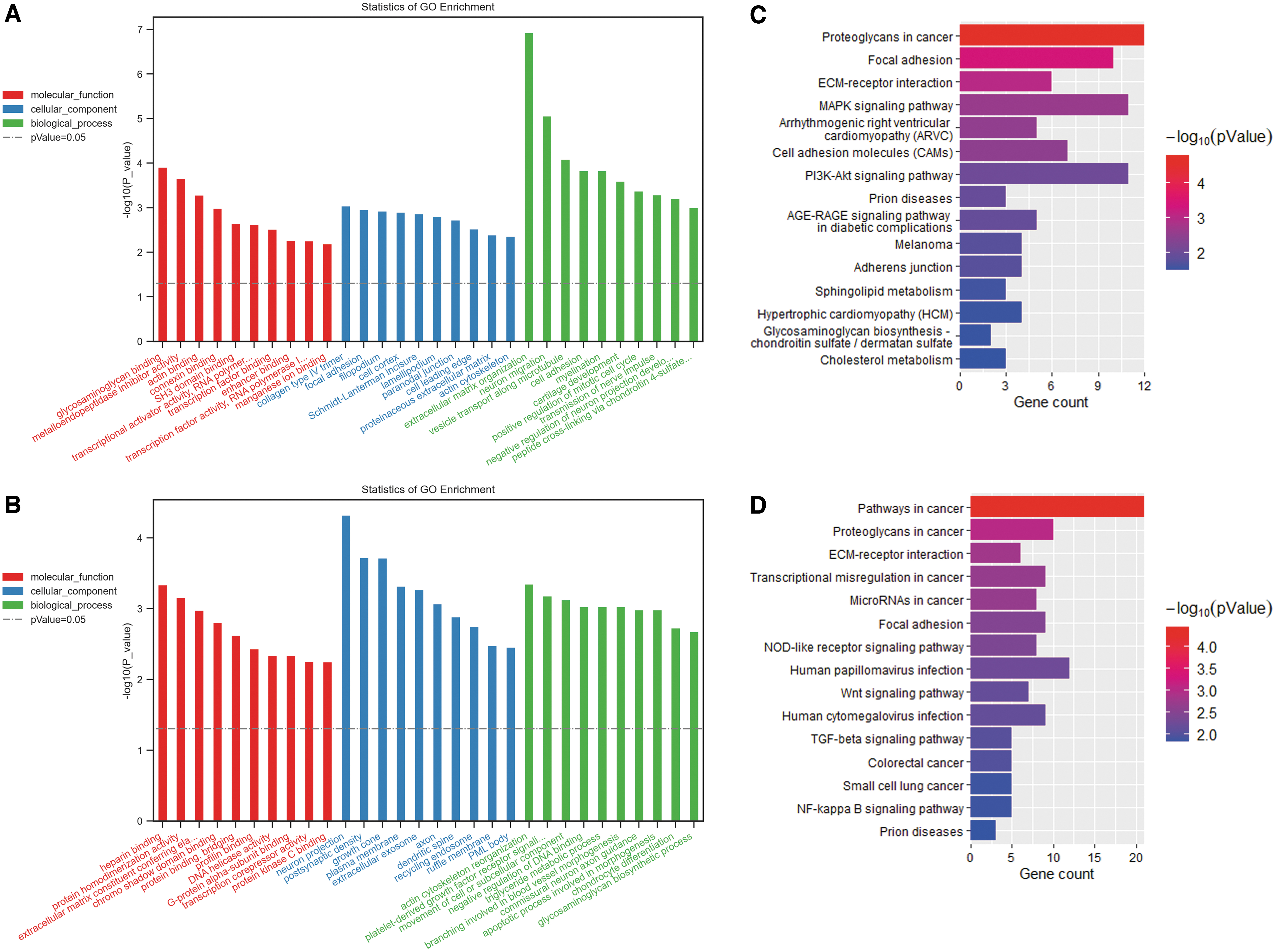
GO function and KEGG pathway analyses of DEcircRNAs-targeted genes.
Identification of hub genes of circRNAs-targeted genes
The list of circRNA-target genes in glomeruli and tubuli was exported to the STRING database, respectively, then PPI networks were constructed. PPI of glomeruli contained 150 nodes and 360 edges (Fig. 7A), whereas tubuli contained 159 nodes and 428 edges (Fig. 7B). Each node represents a protein, and an edge represents an interaction between proteins. The size and gradient color of the nodes were adjusted by the degree. To search for important nodes in the networks, all nodes were ranked by the 12 topological analysis methods provided by CytoHubba. Each algorithm computed all node scores, and then the top 30 nodes were assigned with 1–30 points based on the rank. According to all points, the top 10 nodes in glomerular (CD44, IGF1, DCN, SPARC, HGF, NCAM1, SOX9, NES, and TJP1) (Fig. 7C) and in tubular PPI (CASP3, EGF, CD44, THBS1, DCN, CAV1, CXCL12, RHOA, HSPA5, and THY1) (Fig. 7D) were identified.
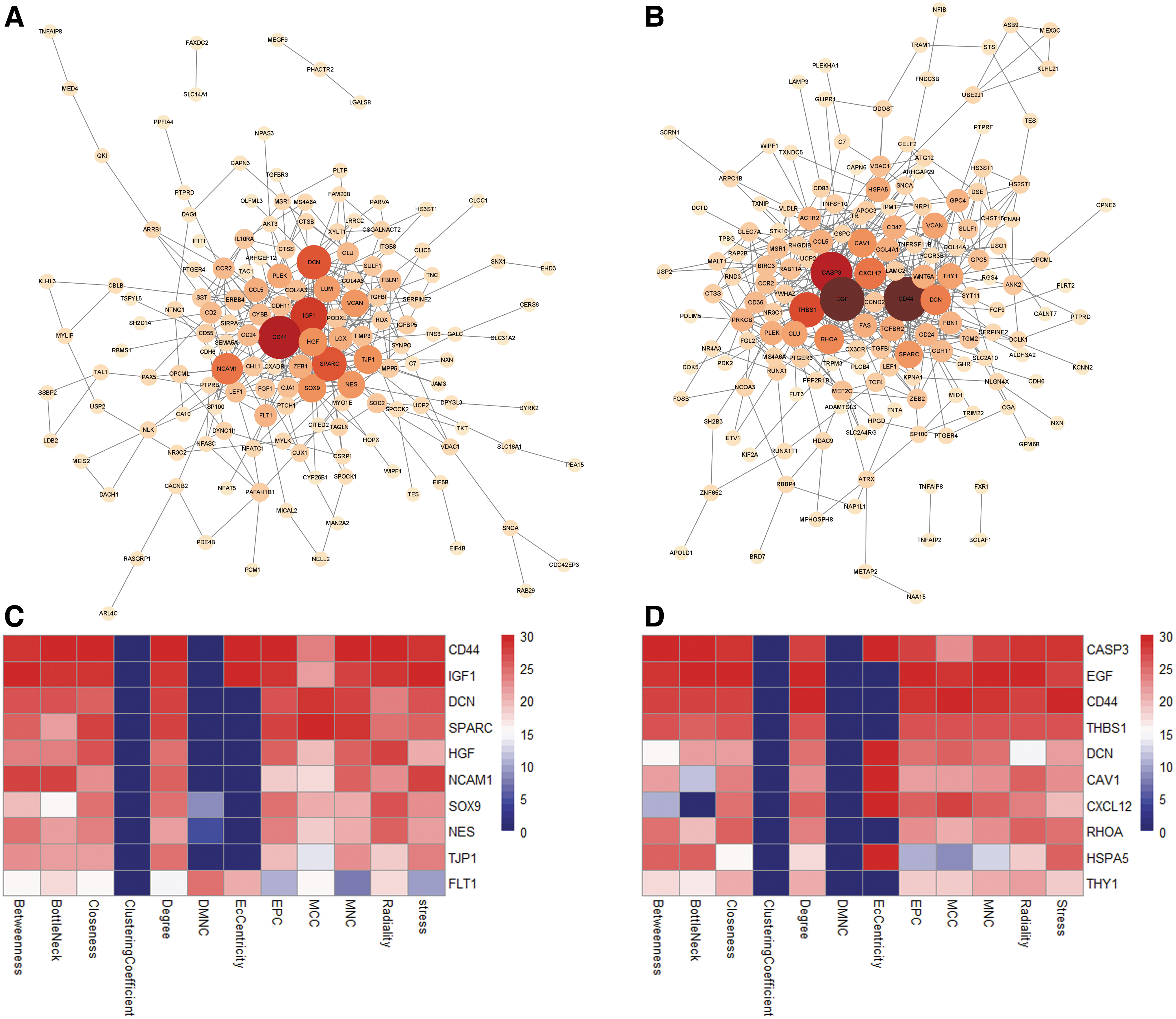
PPI network of circRNA-targeted gene and hub gene identification.
circRNA-targeted hub genes expression was related to renal function
The appearance of proteinuria and change of GFR level are the two main clinical manifestations of DKD, which participate in DKD development. To verify the potential roles of circRNA-targeted hub genes in DKD, renal function data, including GFR of DKD patients and ACR of different DKD mouse models, were obtained from Nephroseq (Woroniecka, Schmid, Hodgin, and ERCB data sets), and Pearson correlation analysis was performed between the hub genes and renal function data. Correlation analysis revealed that the gene expression of HGF and SOX9 in human DKD glomerular samples was negatively related to GFR, whereas TJP1 was positively related to GFR (Fig. 8A). Furthermore, gene expression of SPARC, NES, and CD44 in glomerular samples of different DKD mouse models was negatively related to ACR (Fig. 8B–D). In addition, gene expression of CD44, CAV1, HSP5, CAS3, and RHOA in human DKD tubular samples was negatively related to GFR, whereas EGF and THY1 were positively related to GFR (Fig. 8E–H). All of these data revealed that both in glomerular and in tubular samples, circRNA-targeted hub gene expression was related to renal function of DKD.
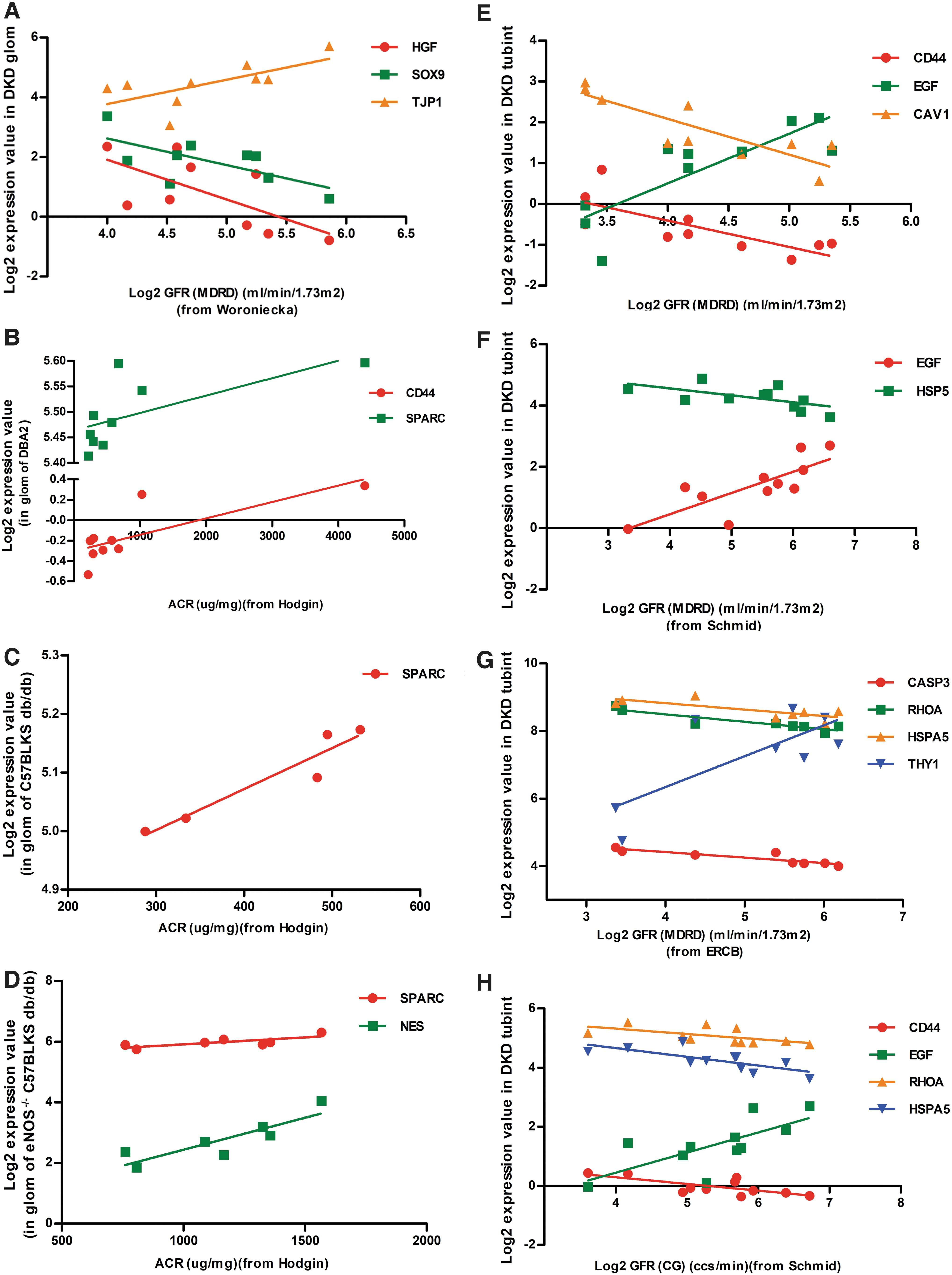
circRNAs-targeted hub genes expression was related with renal function.
Discussion
DKD is characterized by thickening of the glomerular basement membrane, glomerular mesangial hypertrophy and expansion, accumulation of ECM, nodular glomerulosclerosis, arterial hyalinosis, inflammation, and tubular interstitial fibrosis. Among these pathological changes, nodular sclerosis, especially classic Kimmelstein–Wilson nodules, is a highly specific lesion of DKD (Reidy et al., 2014). In this study, mesangial proliferation and expansion, accumulation of ECM, and nodular glomerulosclerosis could be observed in renal tissue of the DKD group, which were consistent with a previous report (Reidy et al., 2014).
Recent study suggests that circRNAs participate in the pathogenesis of many diseases and thus have the potential to serve as potential biomarkers (Lu et al., 2021; Wang et al., 2021). Although some studies about circRNAs profile in db/db mouse or HG-treated cells have been performed (Hu et al., 2019; Peng et al., 2021), the circRNA landscape in kidney of DKD patients has not been reported so far. In our study, there were 63 DEcircRNAs found in the DKD group compared with control, including 40 upregulated and 23 downregulated circRNAs. Considering circRNAs often show cell/tissue and developmental stage-specific expression (Memczak et al., 2013), then we selected 10 DEcircRNAs for validation in HG-treated HMCs and HK2 with qRT-PCR. Results showed that six circRNAs were evidently upregulated in HG-treated HMCs, two circRNAs were obviously upregulated in HG-treated HK2 and four circRNAs were distinctly downregulated in HG-treated HK2, which was consistent with the microarray data.
Because function of circRNA is closely related to its parental gene (Zhang et al., 2013), GO and KEGG pathway analyses of DEcircRNA parental genes were performed. GO analysis revealed that vesicle, focal adhesion, TATA-binding protein (TBP)-class protein binding, single-stranded RNA binding, platelet aggregation, and mitotic spindle assembly were the functions that are most likely regulated by the DEcircRNA. The pathways not only involved focal adhesion, PI3K-Akt signaling pathway, and cell cycle, which might have participated in the pathogenesis of DKD (Gnudi et al., 2016), but also evidently included a variety of cancer disease signals, suggesting that circRNA may take part in the development of DKD and cancer disease through the same signaling pathway, which could provide new window to explore the molecular mechanism of DKD.
Traditionally, DKD is considered to be characterized by pathological changes in glomeruli, but recent studies showed that tubular injury has been increasingly implicated in DKD development and there could be a crosstalk between glomerular and tubular injuries (Slyne et al., 2015; Chen et al., 2020; Vallon and Thomson, 2020). Then, one might ask what is the regulatory role of circRNA in DKD glomeruli and tubuli, respectively, there is no study reported so far. One of important biological functions of circRNAs is to serve as the ceRNA for miRNAs, also known as miRNA sponges (Zheng et al., 2016; Meng et al., 2017; Piwecka et al., 2017). From the perspective of ceRNA, gene expression profile data in glomeruli and tubuli of DKD patients were downloaded from GEO, and ceRNA networks of glomeruli and tubuli were constructed in this study. Furthermore, GO enrichment and KEGG pathway analyses were performed for DEcircRNA targeted gene in ceRNA networks. GO analysis revealed that glomerular genes were mostly enriched in the formation of ECM, such as ECM organization, collagen type IV trimer, and glycosaminoglycan binding, whereas tubular genes evidently enriched in actin cytoskeleton reorganization, neuron projection, and heparin binding. Similar to GO enrichment results, KEGG pathway analysis demonstrated that some glomerular and tubular genes regulated the same signals, whereas some genes mediated distinct pathways, which was consistent with previous reports, that glomeruli and tubuli have different pathophysiological functions but interact with each other and together promote DKD progression (Wang et al., 2016; Warren et al., 2019). Consistent with circRNA parental genes pathway analysis, both glomerular and tubular genes significantly enriched in cancer disease, further suggesting that circRNA probably participated in the progression of DKD and cancer disease by the same signaling pathway.
Bioinformatics analysis revealed that both in glomeruli and in tubuli, circRNA participated in a variety of signal pathways and pathophysiological processes of DKD. Then, whether circRNA could influence renal function of DKD patients? In this study, DEcircRNA-targeted hub genes in glomeruli and tubuli were identified and correlation analyses with renal function were performed, including GFR and ACR, which were the two important clinical indicators of DKD. Results demonstrated that expression of DEcircRNA-targeted hub genes in both glomeruli and tubuli was related with GFR and/or ACR level, again suggesting that both glomerular and tubular circRNA probably regulated renal function of DKD through mechanism of miRNA sponges.
Admittedly, this study has some limitations. First, the number of renal samples in each group was too small, then clinical characteristics of DKD patients and controls could not be statistically analyzed, so further studies should be considered with a larger sample size. In addition, none of the circRNA–miRNA–mRNA bioinformatic predictions have been validated in DKD in vivo or in vitro. In the near future, validation of the identified circRNA–miRNAs–mRNA in the development of DKD will be the focus of our research.
Conclusion
This is the first study to report expression profiles of circRNAs in kidney of DKD patients, and further analysis demonstrated that both in glomeruli and in tubuli, circRNA probably played a significant regulatory role, providing help for understanding the pathogenesis of DKD and investigating novel diagnostic and therapeutic strategies.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Natural Science Foundation of China (Grant Nos. 81770718 and 82000687).
Supplementary Material
Supplementary Figure S1
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.