
Abstract
Ras homologue enriched in brain 1 (Rheb1), an upstream activator of the mechanistic target of rapamycin complex 1 (mTORC1), is known to modulate various cellular processes. However, its impact on bone metabolism in vivo remains unknown. The study aimed at understanding the role of Rheb1 on bone homeostasis. We measured the serum parameters and performed histomorphometry, quantitative real-time polymerase chain reaction, and Western blotting, along with the generation of mouse gene knockout (KO) model, and conducted a microcomputed tomography analysis and tartrate-resistant acid phosphatase staining, to delineate the impacts of Rheb1 on bone homeostasis. In the Rheb1 KO mice, the results showed that Rheb1 KO caused significant damage to the bone microarchitecture, indicating that mTORC1 activity was essential for the regulation of bone homeostasis. Specifically, suppressed mineralization activity in primary osteoblasts and a decreased osteoblast number were observed in the Rheb1 KO mice, demonstrating that loss of Rheb1 led to impaired osteoblastic differentiation. Furthermore, the higher apoptotic ratio in Rheb1-null osteocytes could promote Tnfsf11 expression and lead to an increase in osteoclasts, indicating increased bone resorption activity in the KO mice. The findings confirmed that Rheb1 deletion in osteoblasts/osteocytes led to osteopenia due to impaired bone formation and enhanced bone resorption.
Introduction
Osteoporosis is caused by impaired bone homeostasis, especially in a situation, in which bone-resorbing osteoclasts surpass the bone-forming activity of osteoblasts (Dallas and Bonewald, 2010). The harmony in osteoblast–osteoclast balances the osteoclastic bone resorption and osteoblastic bone formation processes. In this study, the differentiation or activity of cells affects one another. The bone remodeling process allows the skeleton to adapt its structure to the mechanical load by changing its shape, size, and strength (Teti, 2011). Abnormal bone remodeling causes metabolic bone diseases. Therefore, understanding the mechanisms underlying the modulation of bone remodeling is expected to reveal novel strategies for preventing and treating these diseases.
Osteocytes, constituting 90–95% of the total bone cells, are osteoblasts that are embedded in the bone matrix, thereby acquiring a dendritic morphology as well as distinct functions in the specialized lacunae (Salhotra et al., 2020). Osteocytes play a fundamental role in modulating bone remodeling under normal physiological as well as diseased states through their communication via signaling to osteoclasts and osteoblasts in the bone microenvironment (Phillips, 2005). Osteocytes secrete several significant soluble factors that modulate their formation, along with that of the osteoblasts, as well as regulate their functioning. For instance, sclerostin, osteoprotegerin (OPG), and the activator of nuclear factor (NF)-κB ligand (RANKL) act in a coordinated manner to modulate bone resorption along with density, either positively or negatively by regulating the activation state of osteoclasts (Yahiro et al., 2020).
Ras homologue enriched in brain 1 (Rheb1) belongs to the Ras superfamily of GTPases. It is a monomeric protein, ∼21 kDa, functioning as a molecular switch for diverse cellular functions (Heard et al., 2014). Detailed investigations illustrate that Rheb1 serves as a switch that toggles the activity of mechanistic target of rapamycin complex 1 (mTORC1) signaling in the lysosome as a response to the stimuli, thereby controlling anabolism and catabolism (Cornu et al., 2013; Yang et al., 2016). Deletion of Rheb1 impairs mTORC1 activity as demonstrated by decreased S6 phosphorylation in myeloid cells (Li et al., 2017). Rheb also exhibits noncanonical activity, through which its effects seem to be independent of mTORC1 activation (Karbowniczek et al., 2004; Lacher et al., 2010; Xiao et al., 2020). For instance, activated Rheb1 dampens aggresome formation, one centrosome-dependent process, via an mTORC1-independent pathway (Neuman and Henske, 2011).
Moreover, hyperactivation of mTORC1 is implicated in numerous skeletal diseases, including osteoporosis and osteoarthritis (Vasheghani et al., 2015; Ding et al., 2019). Our previous findings in tuberous sclerosis complex 1 (TSC1) knockout (KO) in mature osteoblasts/osteocytes suggest that the activation of mTORC1 reduces sclerostin production and stimulates osteogenesis, resulting in elevated bone mass in the TSC1 gene mutant mice (Liu et al., 2019). However, the role of Rheb1 and the effect of intrinsic impairment of mTORC1 activity on bone homeostasis in mature osteoblasts and osteocytes remain unclear.
In this study, we successfully generated mice with Rheb1-specific KO in mature osteoblasts and osteocytes. Our findings suggested that inhibition of mTORC1 led to osteopenia associated with decreased bone formation and enhanced bone resorption.
Materials and Methods
Experimental animals
All animal experiments were performed with the approval of the Institutional Animal Care and Ethics Committee of Southern Medical University (SYXK 2016-0167). Rheb1 fl/fl mice were a gift from Professor Bo Xiao, Southern University of Science and Technology. The Rheb1 fl/fl and DMP1-cre (JAX stock 023047) mice were mated as described previously (Li et al., 2017; Liu et al., 2019). To generate osteocyte-specific Rheb1 KO mice, first, the DMP1-Cre+ mice were mated with Rheb1 fl/fl mice to obtain DMP1+ Rheb1 fl/+ mice; subsequently, the DMP1+ Rheb1 fl/+ mice were mated with Rheb1 fl/fl mice to obtain the DMP1+ Rheb1 fl/fl line (termed KO) and their littermate control mice (DMP1− Rheb1 fl/fl, termed Con). Mice at 8 and 14 weeks of age were used in this study. Other experimental procedure information is provided in the Supplemental methods (Supplementary Appendix SA1).
Statistics
All quantitative data are presented as mean ± standard error of the mean for a minimum of eight independent samples. Statistical significance in each group was analyzed using the unpaired two-tailed Student's t-test or a one-way analysis of variance (ANOVA).
Results
Generation of mice with osteocyte-specific Rheb1 loss of function
To elucidate the roles of Rheb1 in bone homeostasis, we generated mice lacking Rheb1 expression in mature osteoblasts, as well as osteocytes, by breeding the floxed Rheb1 mice (Rheb1 fl/fl) with DMP1-Cre mice expressing Cre recombinase selectively in osteocytes (Fig. 1A). Results of quantitative polymerase chain reaction (qPCR) (Fig. 1B) and Western blot (Fig. 1C) analyses demonstrated that Rheb1 expression reduced in KO mice relative to the control. This indicated that the mouse model of Rheb1 gene KO was successfully generated. Rheb1 is an upstream activator of mTORC1, and hence, we speculated that when Rheb1 is deleted, the mTORC1 signaling cascade should be inhibited.
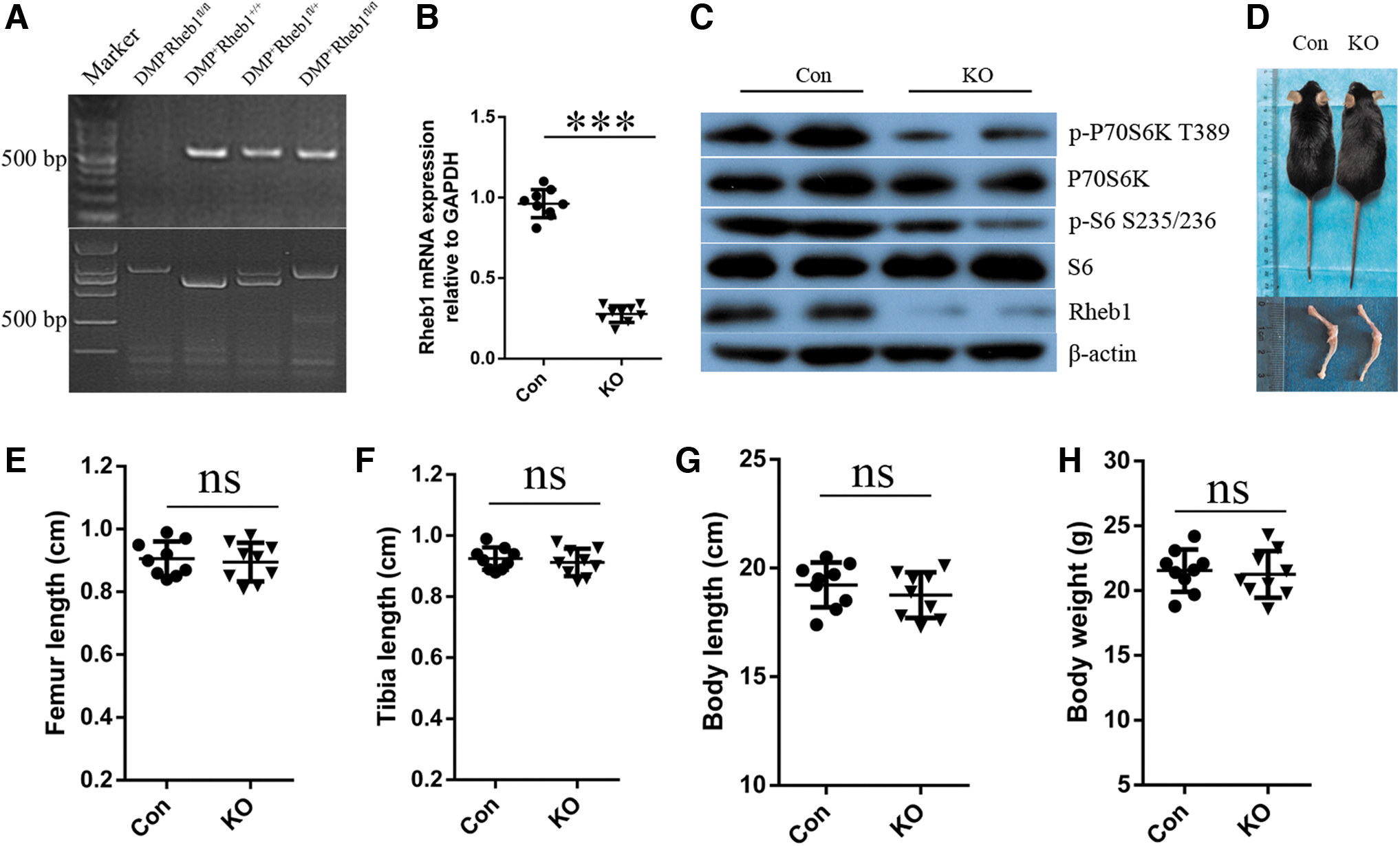
Deletion of Rheb1 in osteocytes did not affect bone growth.
Indeed, the results of Western blot analysis confirmed that p-P70S6K along with p-S6 was inhibited in the KO mice, thereby indicating the inactivation of mTORC1 after Rheb1 disruption in mature osteoblasts and osteocytes. In addition, no significant differences were observed in body weight, body length, and in bone length between KO mice and their control littermates (Fig. 1D–H) matched for age.
Thus, we successfully generated mice with Rheb1-specific KO in mature osteoblasts and osteocytes. Rheb1 deficiency in these cells did not affect the general somatic development.
Rheb1 deletion causes severe osteopenia in mice
To investigate the effects of Rheb1 deletion on bone homeostasis in greater detail, we performed a quantitative assessment of bone microarchitecture using microcomputed tomography (micro-CT) in 8- and 14-week-old male animals. The micro-CT data of the trabecular bone at the distal femur suggested an osteoporotic phenotype in the KO mice (Fig. 2A). In particular, we observed a significant decrease in the bone mass (Fig. 2B), trabecular number (Fig. 2C), and trabecular thickness (Fig. 2D), while an increase was observed in the trabecular separation (Fig. 2E) in KO mice relative to their control littermates. Micro-CT of the midshaft demonstrated that the KO mice indeed showed an obvious phenotype in the cortical bone at 14 weeks relative to their littermate controls (Fig. 2F, G). H&E staining of long bones similarly suggested a substantial decrease in the bone mass in KO mice (Fig. 2H).
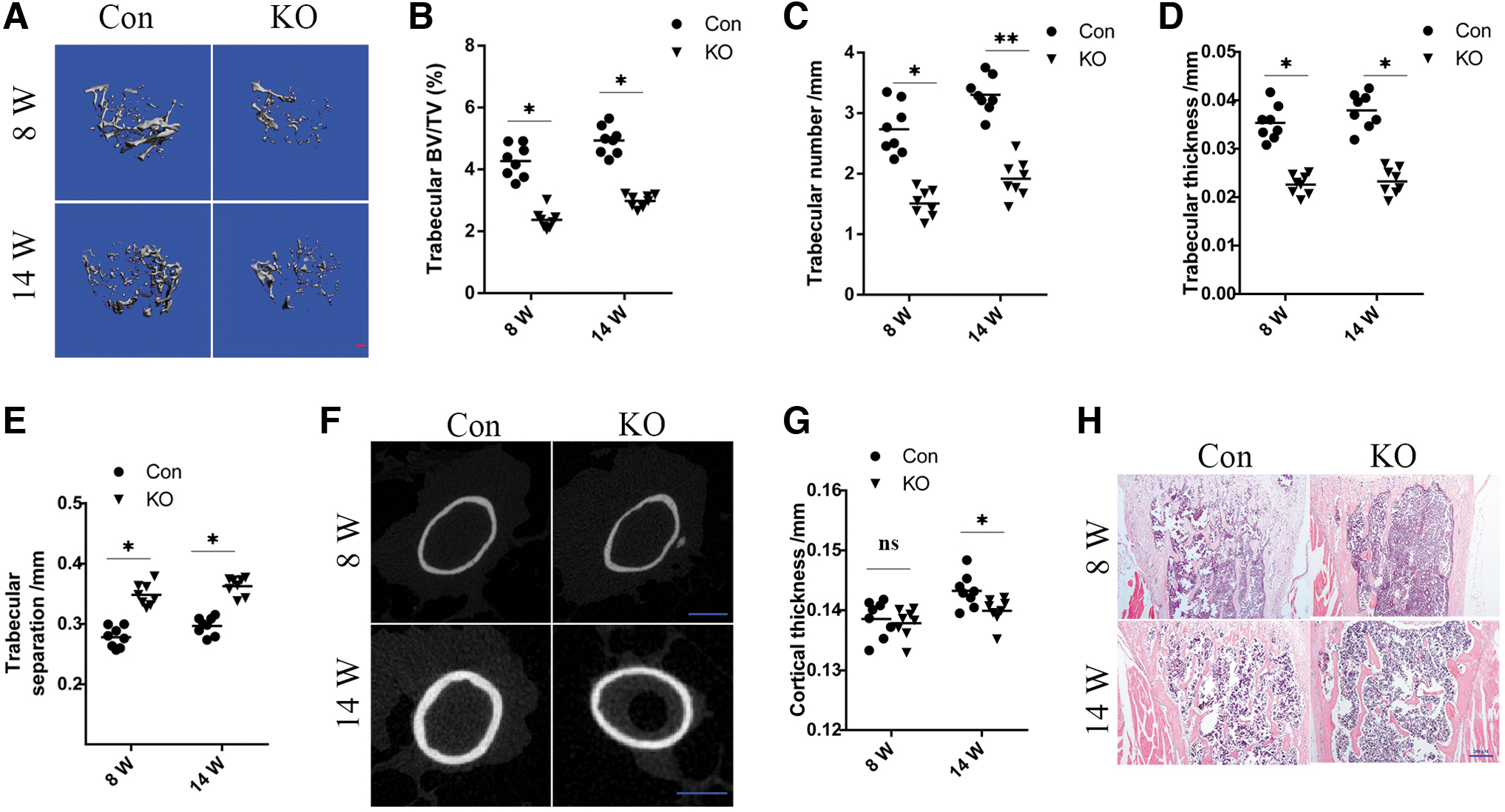
Rheb1 deletion causes severe osteopenia in mice.
Similarly, bone loss was also observed in Rheb1-KO female mice (Supplementary Fig. S1). Thus, the significant decrease in the bone mass observed in Rheb1 conditional KO mice in contrast to their control littermates suggested that Rheb1 affected the function of mature osteoblasts/osteocytes, thereby modulating the bone mass.
Altogether, these data illustrated that Rheb1 deletion in osteocytes led to a meaningful decrease in bone mass of KO mice.
DMP1-Rheb1 mice suffer from severe osteopenia due to diminished bone formation
A reduction in the bone mass was observed in the Rheb1 KO mice, which may be attributed to an increase in osteoclastic bone resorption and/or a reduction in osteoblastic bone formation in the long bone. To examine the mechanisms underlying osteopenia due to Rheb1 loss, diverse sets of experiments were performed. First, we assessed the levels of serum procollagen type 1 amino-terminal propeptide (P1NP), an in vivo biomarker of osteoblast function and bone formation (Moseshvili et al., 2014). Consequently, P1NP was found to be significantly reduced in Rheb1 KO mice relative to the control mice (Fig. 3A). DMP1-cre recombination occurs in osteocytes as well as in the late-stage osteoblast populations (Lu et al., 2016). Indeed, the results of enzyme-linked immunosorbent assay (ELISA) showed that there was a substantial reduction in the levels of osteocalcin in the serum (Fig. 3B), reflective of a reduction in the osteoblast activity in KO mice.
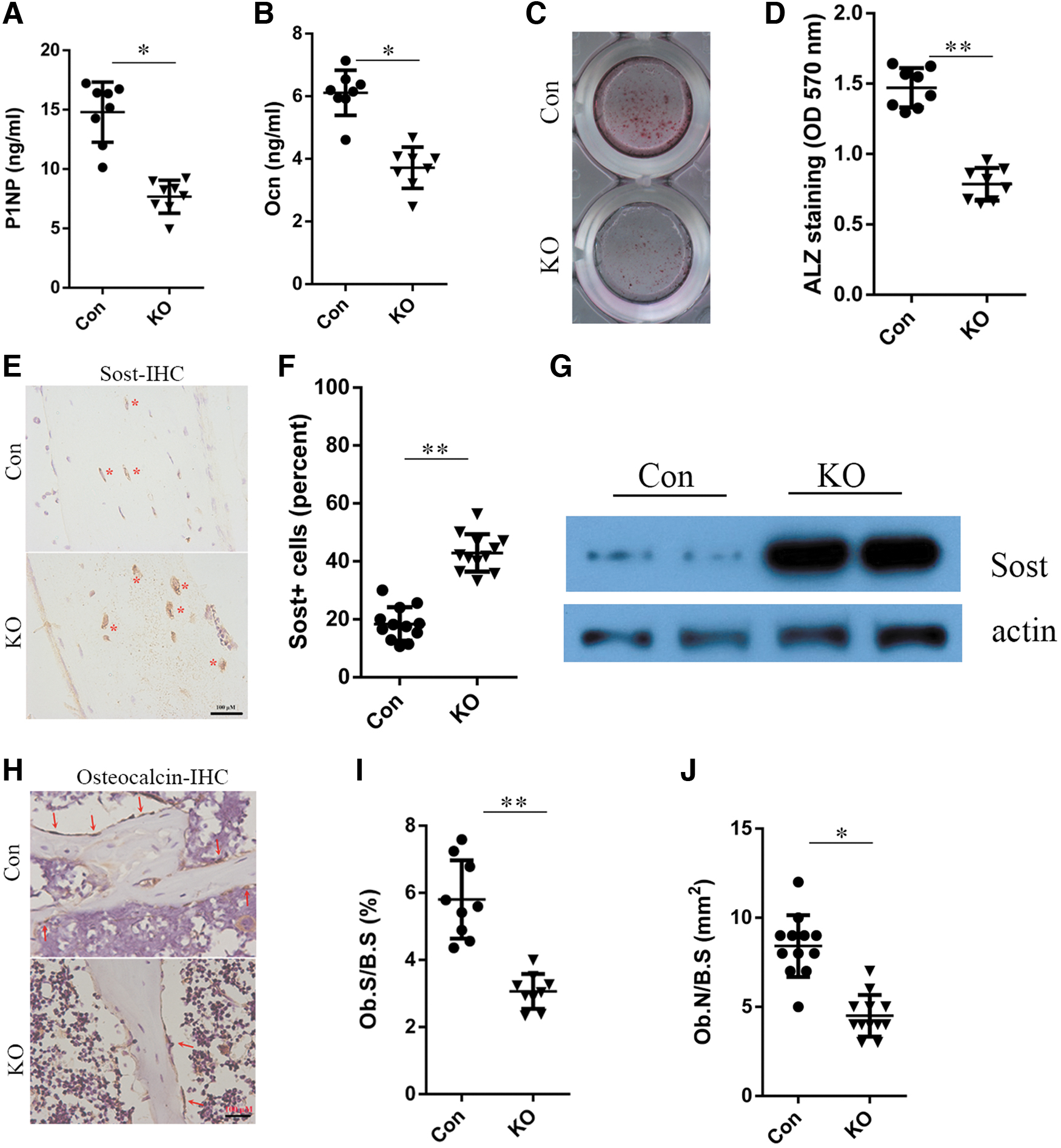
Deletion of Rheb1 in osteocytes decreases bone formation by suppressing osteoblast differentiation.
Moreover, in ex vivo cultures of primary calvarial osteoblasts, we examined whether the bone mass reduction in the Rheb1 cKO mice was due to impaired osteoblast differentiation. From this analysis, the decrease in mineralization activity was considered evidence of attenuated osteogenic differentiation in the KO line relative to the control osteoblasts (Fig. 3C, D). Sclerostin plays an indispensable role in regulating bone mass, and osteocytes are the primary source of sclerostin (Delgado-Calle et al., 2017). Results of immunohistochemistry and Western blotting illustrated that Rheb1 dampened the sclerostin expression in osteocytes, thus reflecting a significant finding in osteocyte biology (Fig. 3E–G). Furthermore, immunohistochemical analyses of the metaphyseal area suggested a decrease in the number of osteocalcin-positive osteoblasts (Fig. 3H–J), thereby implying impaired osteogenesis in the Rheb1 KO mice.
Collectively, these data illustrated that deletion of Rheb1 in mature osteoblasts/osteocytes led to defective bone formation in the KO mice.
Rheb1 deletion increases osteoclast formation and bone resorption
Next, we determined if Rheb1 disruption in mature osteoblasts and osteocytes enhanced the osteoclast activity. Cytochemical staining for tartrate-resistant acid phosphatase (TRAP) is extensively used to identify osteoclasts in vivo (Vaananen and Laitala-Leinonen, 2008). Thus, first, the osteoclast formation in tibial sections from Rheb1 KO and control mice was elevated by TRAP staining. TRAP staining and quantification exhibited a significant increase in osteoclast numbers along with the surface area (Fig. 4A–C) in the secondary spongiosa of Rheb1 KO tibia. C-telopeptide of type I collagen (CTX1) is related to the cathepsin K-dependent cascade and serves as a marker of bone resorption that reflects the functionality of osteoclasts (Henriksen et al., 2007). Furthermore, results of ELISA showed a marked increase in the levels of CTX1 in serum of Rheb1 KO mice relative to the control mice (Fig. 4D).
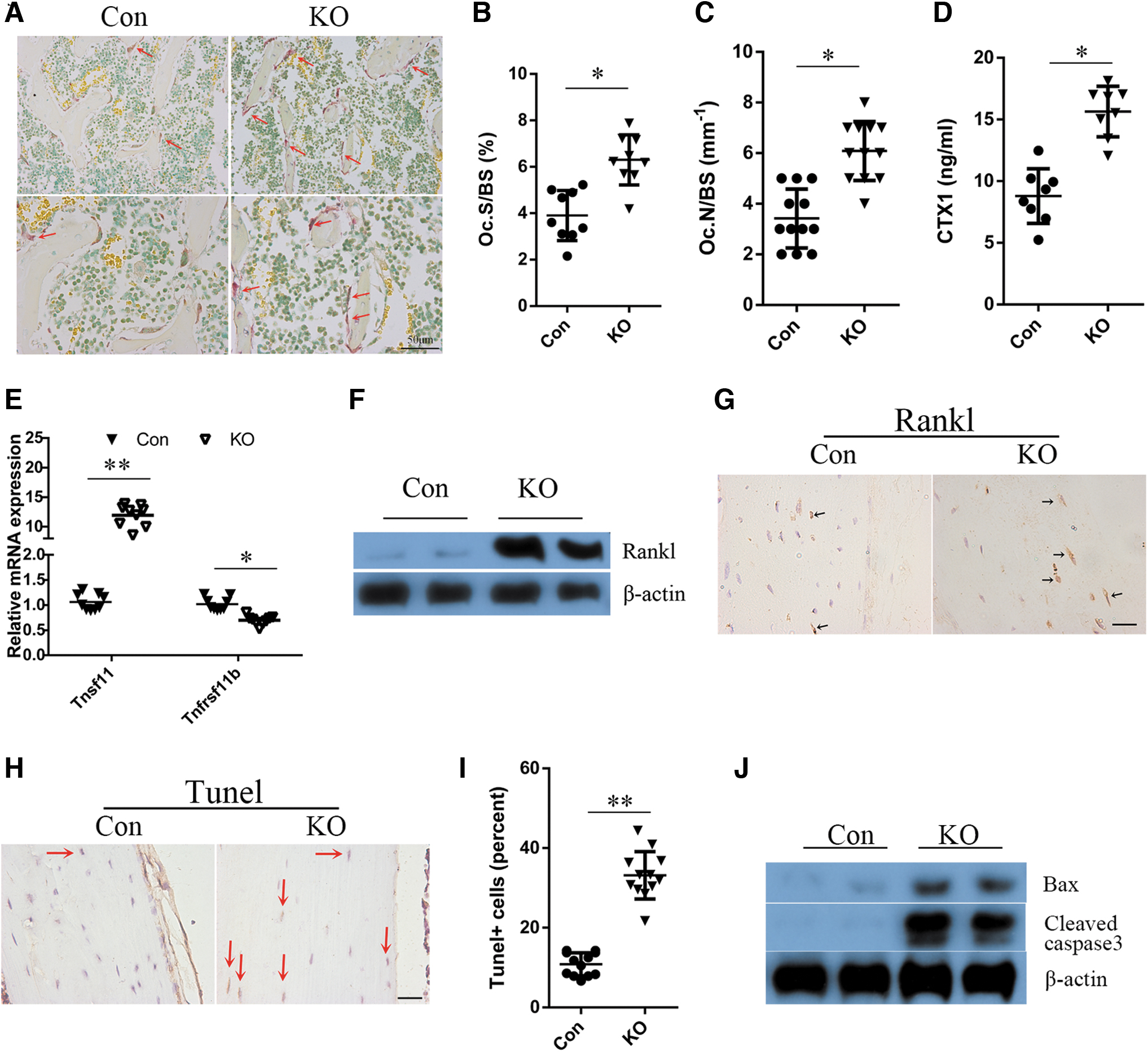
Deletion of Rheb1 in osteocytes increases formation of osteoclasts and resorption of bones.
To investigate whether the activated osteoclast formation in the KO mice was driven by the defective osteoblasts/osteocytes, an ex vivo osteoclastogenesis assay was performed. The control and Rheb1-KO groups showed comparable osteoclast formation after the bone marrow cells (BMCs) were induced by RANKL/M-CSF (Supplementary Fig. S2), which implied that factors secreted from the Rheb1 null osteoblasts/osteocytes may be involved in impaired osteoclastogenesis. Recent breakthroughs have been attributed to the assessment of a family of biologically related proteins, OPG, receptor activator of NF-κB (RANK), and RANKL and modulate osteoclastogenesis. Results of qPCR (Fig. 4E) demonstrated a significantly higher expression of Tnfsf11, and a reduced transcript level of Tnfrsf11b, in the Rheb1 KO mice in contrast to the control littermates.
It has been reported that the 10 kb DMP1-cre, although highly expressed in osteocytes, is also expressed in some mature osteoblasts and BMCs (Fulzele et al., 2018). Thus, we analyzed the levels of expression of Rheb1 and Tnfsf11 in BMCs and cultured osteoblasts from the KO and control mice. As shown in Supplementary Figure S3A, we did not detect any significant differences in Rheb1 expression in the BMCs of control and KO mice, which suggested that the expression of Rheb1 in BMCs of adult KO mice remained unaffected. In a suppressed culture of Rheb1 KO osteoblasts, we detected a decrease in Tnfsf11 expression (Supplementary Fig. S3B). However, the results of Western blot (Fig. 4F) and immunohistochemical staining of tibial sections (Fig. 4G) illustrated an elevation in the protein level expression of RANKL in osteocytes, thereby validating the role of Rheb1-null osteocytes in promoting osteoclast activity.
Apoptotic osteocytes release factors that promote the secretion of RANKL from neighboring osteocytes that influence osteoclast differentiation and bone remodeling (O'Brien et al., 2013). In addition, an elevated apoptosis ratio was observed in Rheb1-KO β cells (Yang et al., 2021). Thus, TUNEL staining was performed and Western blotting was used to investigate the difference in apoptosis rates in control versus KO mice. The percentage of TUNEL-positive cells increased significantly in the KO mice (Fig. 4H, I). Moreover, the protein level expressions of Bax (a proapoptotic factor) and cleaved caspase 3 (an executioner caspase) were increased in the KO mice (Fig. 4J). These data indicated that Rheb1 regulated apoptosis and viability of osteocytes through the mitochondrial apoptosis pathway, which may contribute to the release of RANKL and loss of bone mass in the KO mice.
Collectively, these data highlighted that an increase in osteoclast formation contributed to the low bone mass phenotype in mice with Rheb1 deletion.
Discussion
Based on our findings, we established that the expression of Rheb1 in mature osteoblasts, as well as osteocytes, played a critical role in controlling bone formation. Loss of Rheb1 expression is linked to reduced bone formation along with enhanced bone resorption.
This concept is supported by the following: (a) on the one hand, Rheb1 loss decreased the levels of serum P1NP along with OCN. In, addition, staining suggested inhibition of ALZ in cultured primary Rheb1-null calvarial osteoblasts, along with a reduction in the number of osteoblasts in the Rheb1 KO mice. Furthermore, Western blotting and immunohistochemistry provided direct evidence that Rheb1 loss substantially elevated sclerostin expression in osteocytes, contributing, at least partially, to the dampening of the formation of osteoblasts and repression of the osteoblast function in mutant mice. These data demonstrated that the osteoblast-triggered bone formation activity was dampened upon Rheb1 loss, consistent with loss of bone formation. (b) On the other hand, Rheb1 loss resulted in the marked elevation of the RANKL/OPG ratio, as well as an increase in the protein expression of RANKL in osteocytes, thus promoting the formation of osteoclasts along with bone resorption.
Moreover, the TRAP-positive cell number in the sections of tibial bone in the secondary spongiosa was elevated in the Rheb1 KO mice relative to that in the controls. In addition, there was a significant increase in the level of serum CTX1, as observed in the in vivo osteoclastic bone resorption in Rheb1 KO mice. In addition, the apoptosis ratio was determined in the Rheb1-null osteocytes. Therefore, based on the above findings, we concluded that the deletion of Rheb1 in osteoblasts/osteocytes led to osteopenia, which was associated with reduced bone formation and enhanced bone resorption, and that these changes were a result of osteocyte apoptosis, increases in sclerostin production, and elevated RANKL production.
Rheb1 is a strong activator of mTORC1 signaling cascade when bound to GTP (Heard et al., 2014). mTORC1 consolidates the diverse intracellular along with extracellular signals consisting of growth factors, including stress levels, amino acids, energy, and oxygen to modulate growth of cells, as well as metabolism (Saxton and Sabatini, 2017). The balance between processes of formation and resorption maintains the homeostasis in bones (Kim et al., 2020). The specific deletion of TSC1 in preosteoblasts using Osx-Cre genetically hyperactivated mTORC1 results in osteopenia, primarily because of a defect in bone formation (Huang et al., 2015). A similar phenotype has been reported in mice lacking Raptor expression in preosteoblasts, suggesting that mTORC1 activity is indispensable for the differentiation of osteogenic cells (Shi et al., 2015). Furthermore, repression of mTORC1 with rapamycin impairs osteogenic differentiation of mouse BMSCs ex vivo (Singha et al., 2008), and resulting in loss of trabecular bone in vivo (Xian et al., 2012).
In line with these previous reports, we also observed loss in bone formation in mice lacking Rheb1 expression in mature osteoblasts, as well as osteocytes. Conversely, the deletion of TSC1 in osteocytes was linked to a significant reduction in sclerostin secretion from the osteocytes, increased number of osteoblasts, and subsequently, excessive bone formation (Liu et al., 2019). This was an indication that bone loss in mutant mice was partly due to activated mTORC1-induced suppression of sclerostin. Taken together, these findings were consistent with our conclusions, in that intrinsic mTORC1 signaling could promote bone formation in mice.
In this study, we also established a novel function of Rheb1 in modulating bone resorption through osteocytes. Osteoclasts are responsible for bone resorption, a process pivotal to the maintenance of healthy bones (Mundy and Edwards, 2007). Bone diseases, for instance, osteoporosis, manifest owing to high rates of bone resorption due to osteoclast-induced bone resorption. Recent breakthroughs in the comprehension of differentiation of osteoclasts along with their activation have been attributed to a family of biologically related proteins, including OPG, RANK, and RANKL, which together modulate the formation of osteoclasts.
In a recent study, osteocytes were found to be the primary producers of RANKL in bones and hence were implicated as significant modulators of osteoclastogenesis (Nakashima et al., 2011). Interestingly, Rheb1-mutant mice showed increased expression of RANKL. This increased expression was associated with an increased number of osteoclasts along with enhanced resorption owing to the increased serum level of CTX1, indicating a novel role of Rheb1 in osteocytes in promoting bone resorption.
As the 10 kb Dmp1 promoter is also expressed in some osteoblasts, skeletal muscles, a subset of BMCs, as well as the mammary glands (Dallas et al., 2018), and RANKL is expressed in several tissues, including bone, bone marrow, and mammary ligands (Udagawa et al., 2021), we speculated that Rheb1 deletion in these cell types, may be involved in enhanced osteoclastogenesis. The results of qPCR suggested no significant differences in Rheb1 expression in BMCs, suggesting that the expression of Rheb1 in BMCs of adult KO mice remained unaffected. In a suppressed culture of Rheb1 KO osteoblasts, we detected a decrease in Tnfsf11 expression.
However, Western blotting and immunohistochemical staining of cortical sections illustrated an elevated level of RANKL protein in osteocytes, which directly validated the role of Rheb1-null osteocytes in promoting osteoclast activity. As the expression of Tnfsf11 was reduced in cultured osteoblasts, we speculated that increased osteocyte apoptosis may contribute to the elevation in RANKL production. Studies have demonstrated that osteocyte death was accompanied by large increases in RANKL expression in bone (Tatsumi et al., 2007; Xiong et al., 2011), which may contribute to the increased bone resorption. In our study, we have demonstrated that loss of Rheb1 in osteocyte promotes apoptosis-related protein expression, implying that Rheb1 is a negative regulator of the mitochondrial apoptotic pathway.
In the present study, we have demonstrated the function of Rheb1 in controlling bone mass. In summary, although the precise underlying molecular mechanisms remain to be identified, our findings established that Rheb1 is indispensable in the regulation of bone homeostasis. The results of this study may provide a potential therapeutic target for the prevention and treatment of metabolic bone diseases, including osteoporosis.
Footnotes
Authors' Contributions
J.Y., W.Z., W.L., R.L., P.L., K.Z., and Z.Z. performed the experiments and analyzed the data. J.Y. and X.B. wrote the article. W.W. and X.B. supervised the project and designed the experiments.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants from the National Natural Science Foundation of China (31900507), the Guangdong Basic and Applied Basic Research Foundation (2019A1515011511, 2018A0303130189), the Science and Technology Project of Guangzhou (201904010439), and the China Postdoctoral Science Foundation (2019M652955). The Science Foundation of Shunde Hospital, Southern Medical University (PY2018N108).
Supplementary Material
Supplementary Appendix SA1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.