
Abstract
Together with an anti-tumor immune response, oncolysis using a recombinant viral vector promises to eliminate cancer cells by both gene transfer and host-mediated functions. In this study we explore oncolysis induced by nonreplicating adenoviral vectors used for p14ARF and interferon-β (hIFNβ) gene transfer in human melanoma cell lines, revealing an unexpected role for p14ARF in promoting cellular responses predictive of immune stimulation. Oncolysis was confirmed when UACC-62 (p53 wild-type) cells succumbed upon p14ARF gene transfer in vitro, whereas SK-Mel-29 (p53-mutant) benefitted from its combination with hIFNβ. In the case of UACC-62, in situ gene therapy in nude mice yielded reduced tumor progression in response to the p14ARF and hIFNβ combination. Potential for immune stimulation was revealed where p14ARF gene transfer in vitro was sufficient to induce emission of immunogenic cell death factors in UACC-62 and upregulate pro-immune genes, including IRF1, IRF7, IRF9, ISG15, TAP-1, and B2M. In SK-Mel-29, p14ARF gene transfer induced a subset of these factors. hIFNβ was, as expected, sufficient to induce these immune-stimulating genes in both cell lines. This work is a significant advancement for our melanoma gene therapy strategy because we revealed not only the induction of oncolysis, but also the potential contribution of p14ARF to immune stimulation.
Introduction
Oncolysis, the destruction of cancerous cells in response to treatment, is a clear goal for cancer therapies. Oncolytic viral vectors, for example, selectively destroy tumor cells as a result of virus replication and/or antiviral responses while leaving healthy cells unharmed (Lemos de Matos et al., 2020). The expected advantages to using oncolytic viruses include the amplification of cell killing owing to virus spread as well as the induction of an adaptive immune response against tumor antigens (Lemos de Matos et al., 2020). However, the mechanisms that permit virus replication, in particular reduced type I interferon response in cancer cells, may also limit immune activation (Geoffroy and Bourgeois-Daigneault, 2020).
In our oncolytic gene therapy approach, we have used a nonreplicating adenoviral vector for the transfer of the p14ARF (alternative reading frame of CDKN2a, p14ARF in humans, p19Arf in mice) and interferon-β (IFNβ) genes. We expected p14ARF, a functional partner of p53, to promote cell death, while IFNβ, a pleiotropic cytokine, would contribute to the anti-tumor immune response. Because the vectors did not replicate, there was no concern that IFNβ will disrupt vector function.
In a mouse model of lung carcinoma or melanoma, we have seen that gene transfer of mouse IFNβ (mIFNβ) could induce limited levels of cell killing, but did not provoke immunogenic cell death (ICD) (Catani et al., 2016; Hunger et al., 2017). However, virus-mediated delivery of mIFNβ in association with p19Arf induced ICD as verified by emission of danger-associated molecular patterns (DAMPs) as well as stimulation of anti-tumor immune responses in vivo (Catani et al., 2016; Medrano et al., 2016; Hunger et al., 2017). We also confirmed these results using an ex vivo model of human T cell priming (Cerqueira et al., 2020). Thus, combined gene transfer using a nonreplicating vector induced oncolysis accompanied by ICD.
A recent study has shown that pharmacological inactivation of MDM2/MDMX results in p53-dependent induction of immune-stimulatory factors (Zhou et al., 2021). In the work of Zhou et al., inhibition of MDM2/MDMX by Nutlin-3 resulted in decreased DNA methylation, leading to expression of endogenous retroviruses which, in turn, activated antiviral and pro-immune responses in tumor cells bearing wild-type p53. A new study has shown the p53 can induce ICD when delivered by an oncolytic adenovirus in a model of pancreatic cancer (Araki et al., 2022). Since the primary activity of p14ARF is the liberation of p53 from MDM2 (Zhang et al., 1998; Tao and Levine, 1999; Weber et al., 1999), we explored whether p14ARF gene transfer would activate the expression of immune-stimulating genes, which, in turn, may improve tumor elimination.
In this study we used adenoviral vectors where transgene expression is under the control of the constitutive cytomegalovirus (CMV) promoter, thus permitting the application of these vectors to a variety of cells, including those with mutant p53, an important point because our previous studies used vectors where transgene expression was controlled by a p53-responsive promoter, thus limiting expression to cells that harbor wild-type p53 (Catani et al., 2016; Medrano et al., 2016; Hunger et al., 2017; Cerqueira et al., 2020). We tested individual and combined p14ARF and human IFNβ (hIFNβ) gene transfer as a means to induce oncolysis, ICD, and inhibit tumor progression in human melanoma cells, revealing a previously unknown role for p14ARF in promoting factors that predict an anti-tumor immune response.
Materials and Methods
Cell culture
The human embryonic kidney (HEK293) cell line (Thermo Fisher Scientific, Waltham, MA) and the human melanoma cell lines UACC-62 and SK-Mel-29 (kindly provided by Roger Chammas, ICESP-FMUSP, authenticated using STR profiling and routinely tested negative for mycoplasma by a PCR assay) were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 1 × antibiotic-antimycotic and 10% fetal bovine serum (FBS) (all from Thermo Fisher Scientific) at 37°C, 5% CO2.
Vector construction
For the generation of the recombinant adenovirus, the fiber-modified adenovirus plasmid (pAdHM15-RGD, kindly provided by Dr. Hiroyuki Mizuguchi, Osaka University, Japan) (Mizuguchi et al., 2001) was modified to be compatible with the Gateway site-directed recombination strategy. For this, the “destiny” sequence necessary for recombination was removed from pAAV-Dest (Thermo Fisher Scientific) and inserted in pShuttle2 (Takara Bio, Mountain View, CA).
The cassette comprising the CMV promoter plus the “destiny” recombination sequence was inserted in pAdHM15-RGD, creating pAdRGD-CMV/dest, as per a similar, previously described procedure (Hunger et al., 2017). The transgenes were cloned into the EcoRI site of the pENTR 2B plasmid (Thermo Fisher Scientific) and then transferred to pAdRGD-CMV/dest by homologous recombination utilizing Gateway L/R Clonase II Enzyme (Thermo Fisher Scientific), giving rise to AdRGD-CMV-Luc2, AdRGD-CMV-Luc2-P2A-p14ARF, and AdRGD-CMV-Luc2-T2A-hIFNβ. Additional information regarding cloning is available upon request. The AdRGD-CMV-LacZ vector was provided by Dr. Mizuguchi (Mizuguchi et al., 2001).
Adenovirus rescue and quantification
Virus production started by transfecting the HEK293 cells with the plasmids digested with PacI enzyme (New England Biolabs, Ipswich, MA). After subsequent steps of amplification, the viruses were purified by iodixanol gradient centrifugation, as described elsewhere (Peng et al., 2006; Hunger et al., 2017) and titers obtained through biological quantification using the Adeno-X Rapid titration kit (Takara Bio). This biological titer, expressed as transducing units (TU)/mL, was used in calculating the multiplicity of infection (MOI) for each assay.
Cell transduction
The transduction procedure was designed to guarantee a consistent number of viral particles and gene dosage, as outlined in Supplementary Table S1. UACC-62 and SK-Mel-29 cells were seeded in a minimal volume of media containing 2% FBS in the presence of virus (total MOI of 200, unless otherwise noted) of the corresponding vector/s, incubated at 37°C overnight and then fresh medium containing 10% FBS was added, incubation continued, and cells were evaluated as described for each assay.
LacZ
For each cell line, 1 × 105 cells/well were plated in six-well dishes and transduced with the indicated MOI using AdRGD-CMV-LacZ. After 24-h incubation, the cells were fixed (1 × phosphate-buffered saline [PBS], 2% paraformaldehyde, 0.2% glutaraldehyde) at 4°C for 5 min and then stained to reveal β-galactosidase activity [3 mM K3Fe(CN)6, 3 mM K4Fe(CN)6, 1.2 mM MgCl2 and 1 mg/mL X-gal in 100 mM sodium phosphate buffer, pH 7.3], 37°C, overnight. Random fields, 10/condition, were photographed and total and blue-stained cells were counted to calculate the percentage of cells with β-galactosidase activity and, thus, adenovirus transduction.
Luciferase detection
Cells were transduced, incubated at 37°C for 48 h, harvested, and then luciferase activity was assessed using the Luciferase Assay System kit (Promega, Madison, WI) as per the manufacturer's protocol. Luminescence was read on a 1420 Multilabel Counter VICTOR3 (Perkin Elmer, Waltham, MA). The luminescence results were normalized by protein concentration.
Interferon-β detection
Cells were transduced, incubated at 37°C for 48 h before cell culture supernatant was collected, and the levels of secreted hIFNβ were assessed using Verikine human IFN beta ELISA kit (PBL Assay Science, Piscataway, NJ).
Immunofluorescence for p14ARF
After 48-h incubation, transduced cells were fixed with cold 4% paraformaldehyde, 4°C for 20 min, and then treated with PBS—nonidet P40 0.1% for 30 min at 37°C. Samples were incubated with the primary mouse IgG anti-p14ARF antibody (sc-53640; Santa Cruz Biotechnology, Santa Cruz, CA) followed by the secondary AlexaFluor 594 anti-mouse IgG (Thermo Fisher Scientific). Immunofluorescence detection was obtained using EVOS FL Cell Imaging System (Thermo Fisher Scientific).
Quantitative PCR
Cells were transduced as indicated in Figures 1 –8. Alternatively, cells were incubated in the presence of 10 μM Nutlin-3 (Sigma-Aldrich, St. Louis, MO) for 24 h before collecting cells for analysis. Oligonucleotide sequences are listed in Supplementary Table S2. Total RNA was extracted using TRIzol™ reagent (Thermo Fisher Scientific) as recommended by the supplier before performing quantitative PCRs (qPCRs) using GoTaq® 1-Step RT-qPCR System (Promega) and 7500 Real-Time PCR System (Thermo Fisher Scientific) as recommended by the supplier. Expression levels were analyzed using the ΔΔCt method. GAPDH mRNA level was used as an internal control.
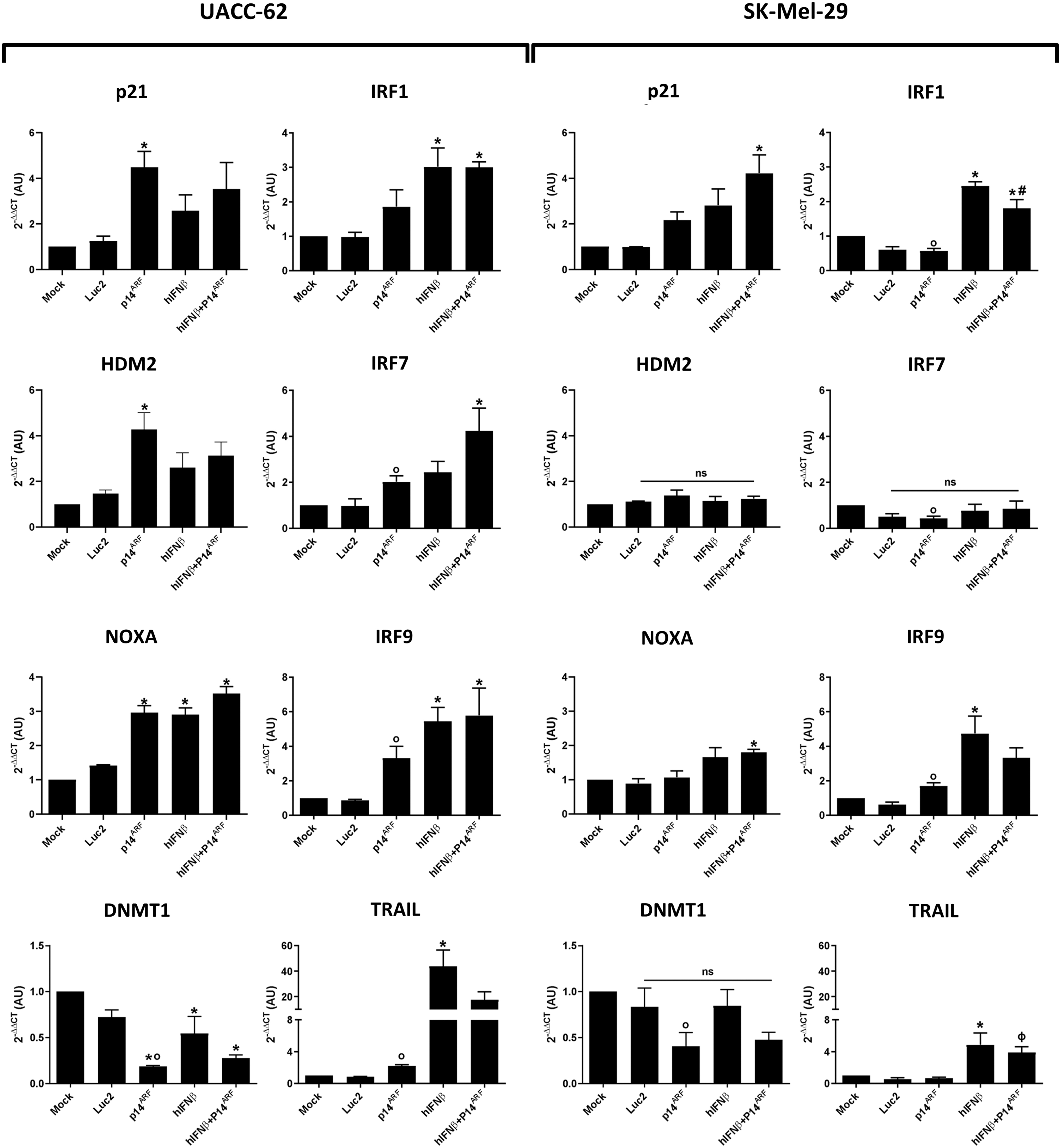
Transgene activity revealed by qPCR analysis of transcripts of key target genes in the p53 and interferon pathways. UACC-62 and SK-Mel-29 cells were mock transduced or transduced at a MOI of 200 (total) as given in Supplementary Table S1 with AdRGD-CMV-Luc2 (Luc2), AdRGD-CMV-Luc2-P2A-p14ARF (p14ARF), AdRGD-CMV-Luc2-T2A-IFNβ (hIFNβ), or the combination (hIFNβ+p14ARF) and incubated for 48 h before recovery of total RNA and reverse transcription qPCR analysis. GAPDH was used as the reference gene. *p < 0.05 versus Mock, ANOVA one-way, Bonferroni post-test. ϕ p < p14ARF+hIFNβ versus p14ARF. ANOVA one-way, Bonferroni post-test. # p < 0.05 p14ARF+hIFNβ versus p14ARF and hIFNβ, ANOVA one-way, Bonferroni post-test. o p < 0.05 versus Mock, unpaired t-test. ns, not significant. N = 3–4 biological assays. ANOVA, analysis of variance; CMV, cytomegalovirus; MOI, multiplicity of infection; qPCR, quantitative PCR.

Impact of individual and combined gene transfer on the population dynamics of melanoma cell lines.
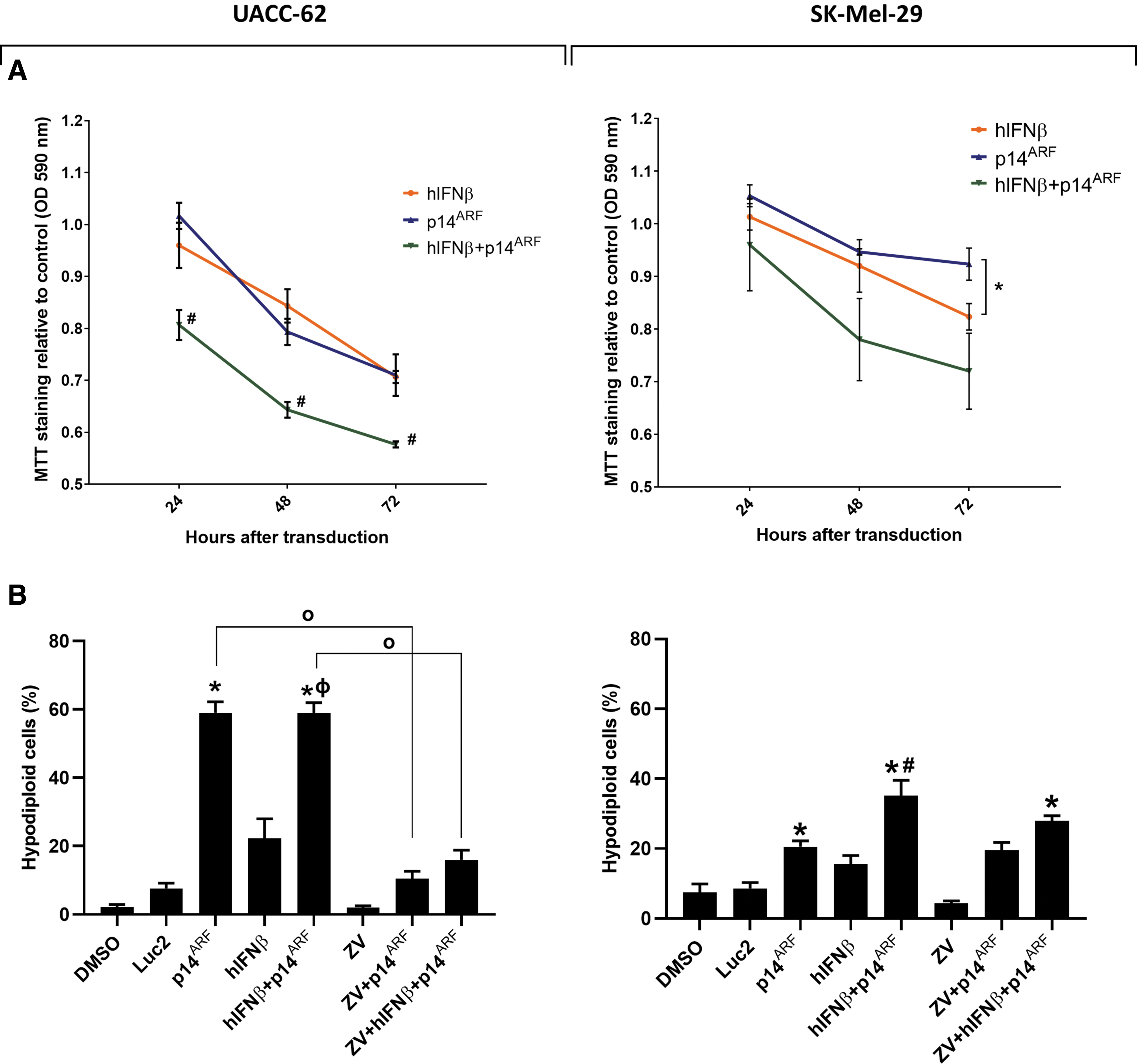
Viability of melanoma cell lines after individual and combined p14ARF and hIFNβ gene transfer.
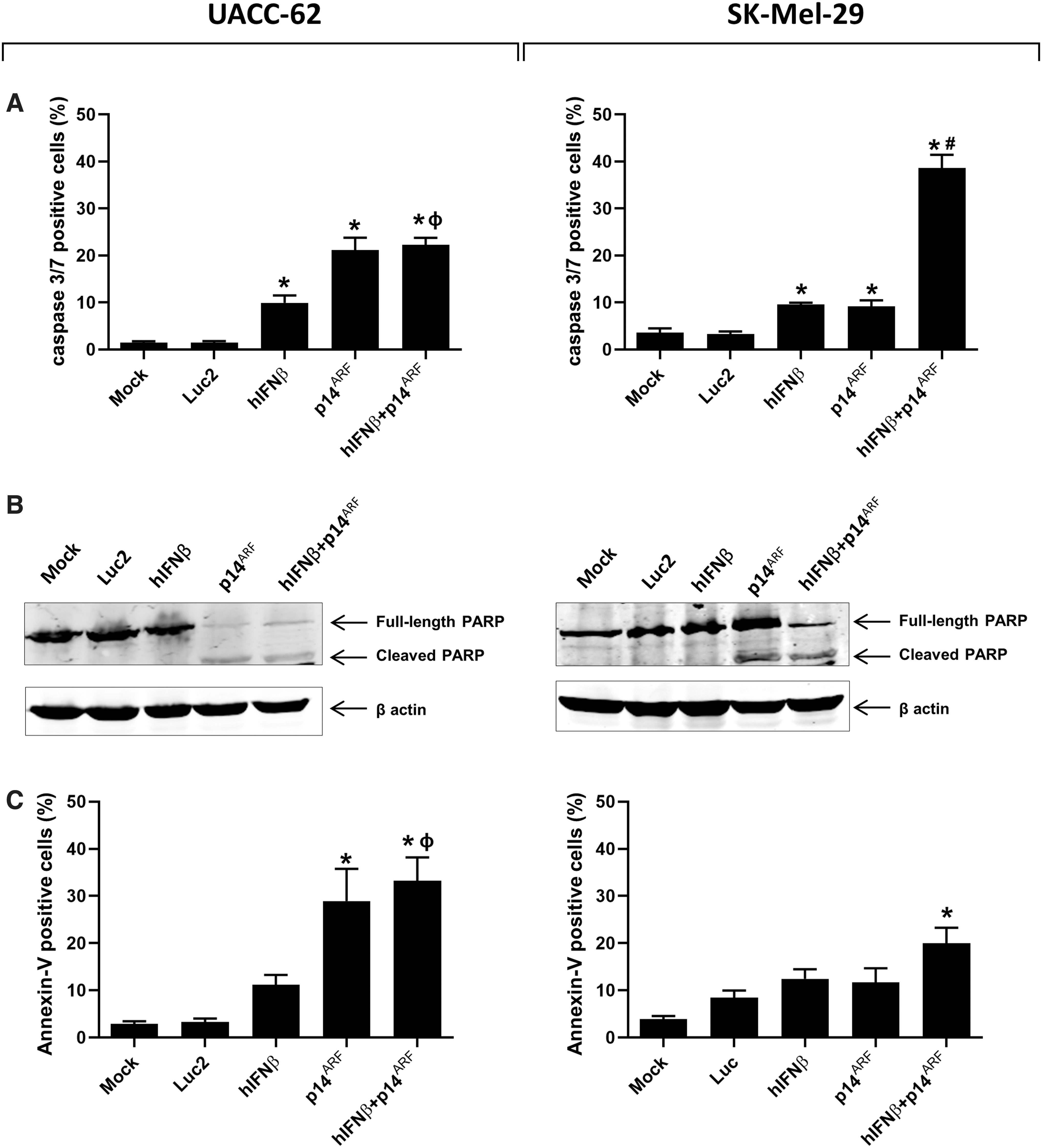
Cell death mechanism investigated upon individual and combined gene transfer of p14ARF and hIFNβ in melanoma cell lines.
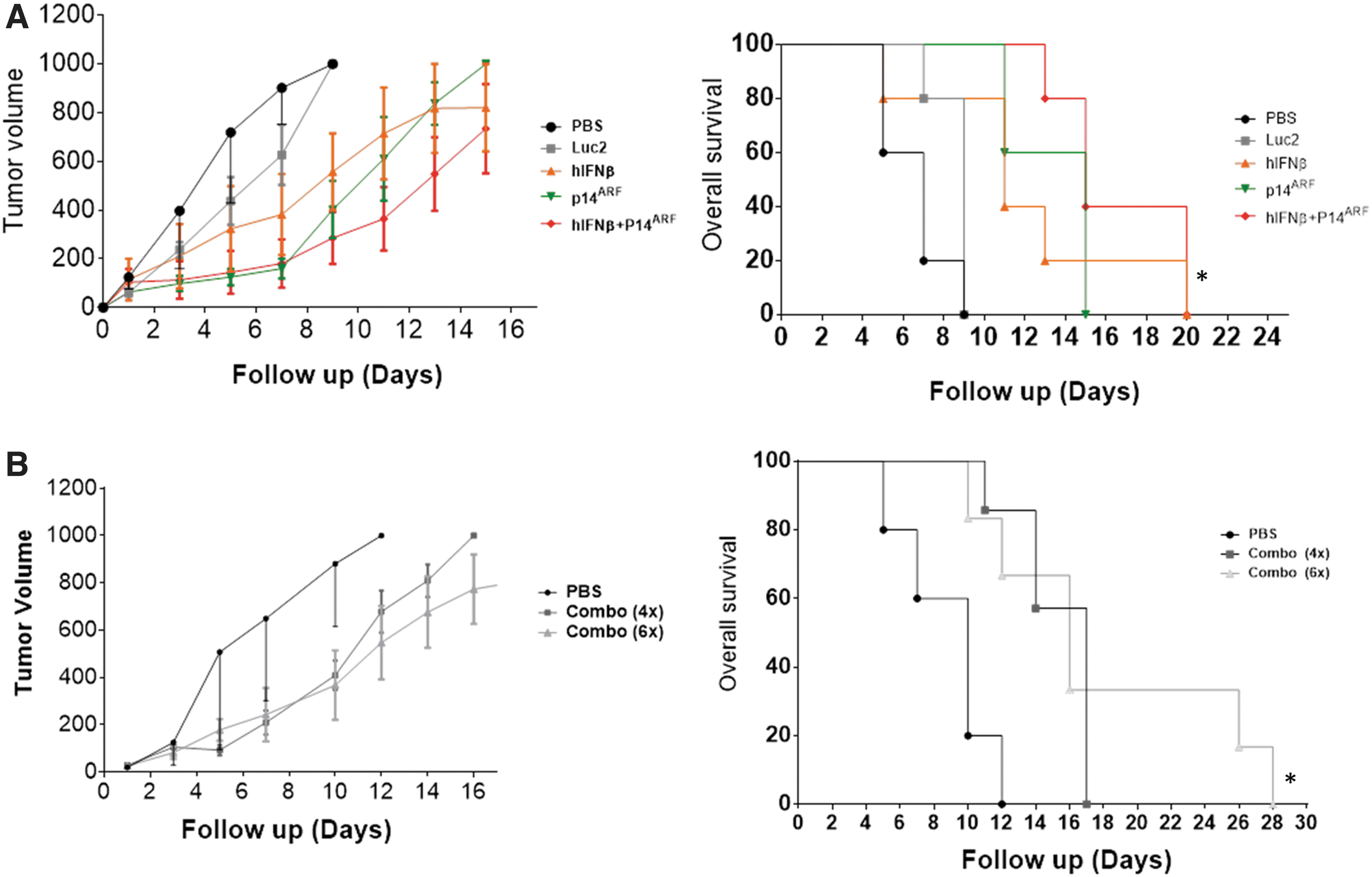
In situ gene therapy. UACC-62 cells were injected s.c. in nude mice and tumors were allowed to form before intratumoral injection with the indicated adenoviral vectors and posterior observation.
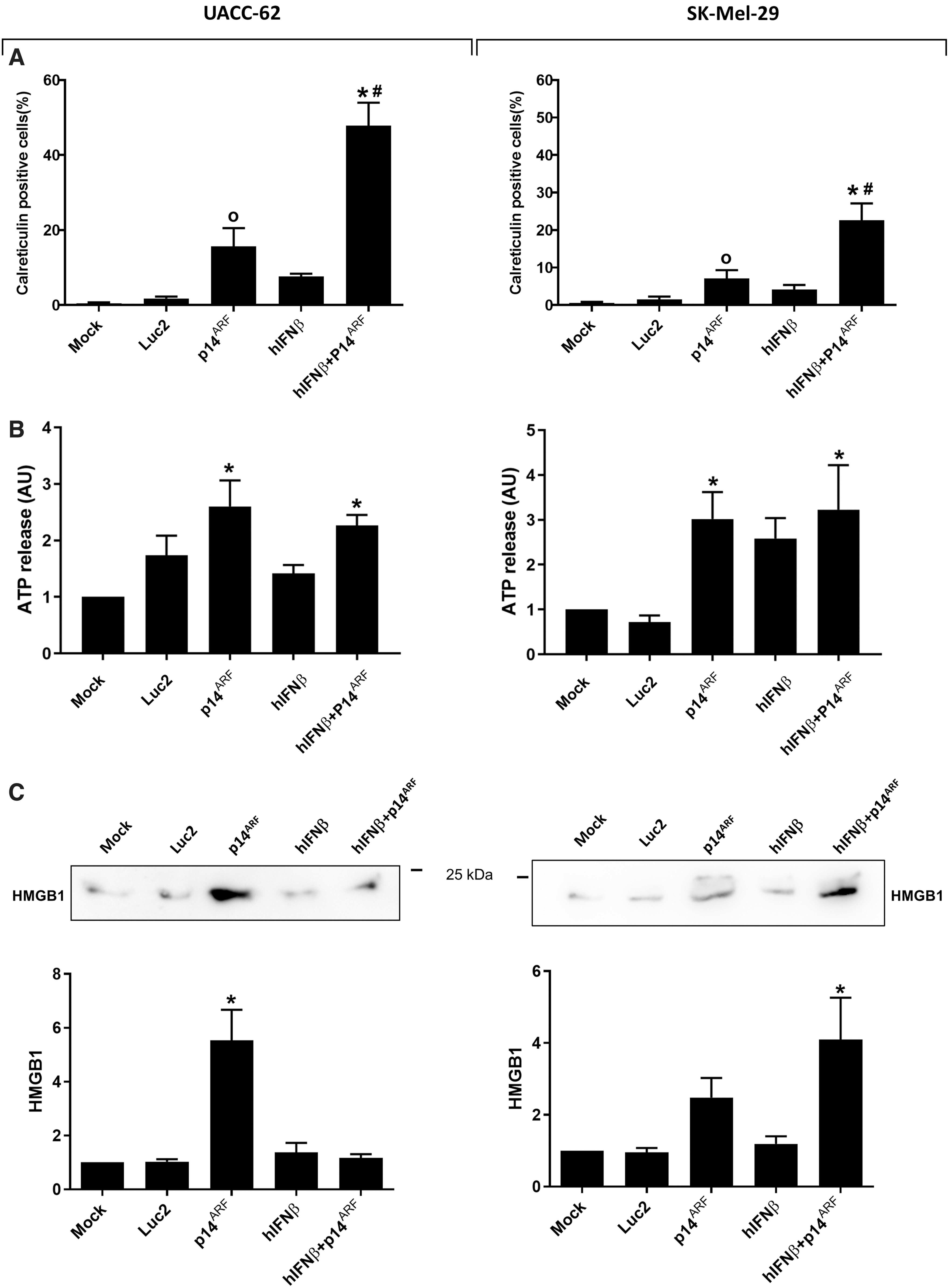
Emission of ICD markers upon gene transfer in melanoma cell lines. Cells were mock transduced or transduced with the indicated vectors and, 48 h after, the cells were harvested and analyzed for the emission of ICD markers.

Upregulation of immune-stimulatory genes upon p14ARF gene transfer. Cells were mock transduced or transduced with the p14ARF adenovirus and incubated for 48 h or treated with 10 μM Nutlin-3 and incubated for 24 h before recovery of total RNA and reverse transcription qPCR analysis. GAPDH was used as the reference gene. *p < 0.05 versus mock, ANOVA one-way, Bonferroni post-test. # p < 0.05 p14ARF versus Nutlin-3, ANOVA one-way, Bonferroni post-test. ns, not significant. N = 3–4 biological assays.

Detection of RIG-I, ISG15 and B2M at the protein level. Cells were mock transduced or transduced with the p14ARF adenovirus and incubated for 48 h or treated with 10 μM Nutlin-3 and incubated for 24 h before protein analysis.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide and colony-formation assay
To assess cytotoxicity, 5 × 103 cells were seeded and treated with the vectors. After 24-, 48-, and 72-h incubation after transduction, the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test was carried out following the previously described protocol (Tamura et al., 2016). For the clonogenic assay, 5 × 104 cells were treated as already mentioned. The next day, cells were recovered with trypsin, washed in PBS, and 1 × 104 cells were seeded in 100 mm culture dishes and then incubated at 37°C. After 12 days, cells were washed with PBS and stained with 2% crystal violet solution in ethanol.
Cell growth curve
Here, 5 × 104 cells were transduced following the described protocol and after 24-, 48-, and 72-h incubation at 37°C, adherent cells were recovered, stained with trypan blue, and the number of viable (unstained) cells was determined by manual counting.
Flow cytometry for hypodiploid population assessment
One hundred thousand cells were treated with the vectors and after 72-h incubation, both detached and adherent cells were recovered and fixed in 70% ethanol followed by RNAse treatment and staining with propidium iodide (PI). Flow cytometry results were obtained using Attune flow cytometer (Thermo Fisher Scientific).
Phosphatidylserine exposure and caspase 3/7 activation
For both assays, 1 × 105 cells were treated with the vectors, incubated for 48 h and then both floating and attached cells were recovered, washed once in 1 × PBS, and then stained. For the annexin V assay, cells were stained with Annexin V–Alexa 488 (Thermo Fisher Scientific) and PI, following the manufacturer's protocol. To assess caspase activation, cells were treated with CellEvent Caspase 3/7 detection reagent (Thermo Fisher Scientific) according to the protocol provided. Flow cytometry results were obtained using the Attune flow cytometer (Thermo Fisher Scientific).
Western blot detection of PARP
Two hundred thousand cells were treated with the vectors in 12-well plates, incubated for 48 h and then total protein extract was obtained and submitted to western blot, as previously described (Merkel et al., 2013). After blocking in the presence of 5% dry milk, the membranes were probed with the rabbit anti-PARP-1 [Poly(ADP-ribose) Polymerase] (Cat. No. 9542; Cell Signaling, Danvers, MA) and mouse anti-β-actin (A5441; Sigma Aldrich) primary antibodies, followed by fluorescent-coupled secondary antibodies (LI-COR Bioscience, anti-rabbit Ref. 926-32223, anti-mouse Ref. 926-32212). Bands were detected using the Odyssey near-infrared image system (LI-COR Biosciences).
Z-VAD-FMK caspase inhibition assay
Cells were first transduced with the vector, incubated for 20 h, and then the media were replaced with DMEM/10% FBS containing either the pan caspase inhibitor Z-VAD-FMK (Santa Cruz Biotechnology) at 20 μM or dimethyl sulfoxide and the cells were then incubated for an additional 28 h (48-h total post-transduction). Then both floating and adherent cells were harvested, fixed in 70% ethanol, washed with PBS, and then PI stained, as described previously, for detection of hypodiploid cells. Results were obtained using the Attune flow cytometer (Life Technologies).
ICD markers
The ICD markers calreticulin and ATP were analyzed following previously published protocols (Catani et al., 2016; Hunger et al., 2017). For each assay, 1 × 105 cells were treated with the vectors, incubated for 48 h and then cells and supernatant were collected. For the evaluation of calreticulin exposure through flow cytometry, cells were probed with rabbit anti-calreticulin antibody (NB300-545; Novus Biologicals, Littleton, CO), followed by the Alexa488-conjugated anti-rabbit secondary antibody (Thermo Fisher Scientific). The cells were then analyzed using the Attune flow cytometer (Thermo Fisher Scientific).
Secreted ATP was evaluated in conditioned medium using the ENLITEN ATP Assay System (Promega), following the manufacturer's protocol, and the luminescence obtained with a GloMax Plate Reader (Promega).
HMGB1 was measured by western blot where conditioned cell culture media was centrifuged 1 min at 12,000 g and supplemented with protease inhibitor cocktail (Thermo Fisher Scientific). From this, 180 μL of media were concentrated (Concentrator Plus; Eppendorf, Hamburg, Germany) and submitted to western blotting and detection using anti-HMGB1 (1:1000, ab79823; Abcam, Cambridge, United Kingdom) and a horseradish peroxidase–conjugated secondary antibody (1:10,000; Sigma Aldrich) visualized with ECL (GE Healthcare, Chicago, IL). Images were recorded using an ImageQuant LAS4000 imaging platform (GE Healthcare).
In vivo assays
All animal experimentation and protocols described in this study were approved by the Ethics in Animal Use Committee (CEUA) of the Faculdade de Medicina, Universidade de Sao Paulo (FMUSP), Protocol No. 167/14, and performed at the Centro de Medicina Nuclear (CMN), FMUSP, São Paulo, Brazil. UACC-62 cells (1 × 106) were implanted subcutaneously in the flank of the athymic male nude BALB/c mice, 8–12 weeks old. The condition of these animals, as well as tumor growth, was monitored three times per week. Tumors were measured with calipers and the volume calculated according to the formula: ½ × (length) × (width)2 (Tomayko and Reynolds, 1989; Tamura et al., 2019). When tumors reached 60 mm3, treatment was initiated with 2 × 108 TU of adenovirus in 50 μL 1 × PBS/injection delivered intratumorally. This treatment was repeated a total of four or six times, as indicated, at 48-h intervals (days 1, 3, 5, 7, 9, and 11).
We consider the tumor volume of 1000 mm3 as the experimental end point when the animals were anesthetized in a chamber with 4% isoflurane and killed by CO2 inhalation. Animals that did not reach the end point were monitored until 60 days. Distribution of animals/cage was maintained according to the predefined experimental groups at the beginning of the treatment. For the assay given in Figure 5A, N = 5/group (total of 25 animals); in Figure 5B, N = 5 (PBS), N = 7 (Combo 4 × ), N = 6 (Combo 6 × ), for a total of 18 animals. These assays were performed only on the occasion described in this study.
Immunohistochemistry
Tumors were recovered 48 h after the final (fourth) treatment, fixed in formalin, embedded in paraffin, and 5 μm sections were mounted on silanized microscope slides. Immunohistochemistry was carried out as previously published (Xande et al., 2020). The primary antibody (anti-Ki67, ab15580; Abcam, Cambridge, MA) was diluted 1:50 in PBS containing 5% serum. The Vectastain universal biotinylated secondary antibody was diluted 1:100 in PBS containing 5% serum. The images used for analysis were captured with light field microscopy using a 20 × objective. Five images of each animal were randomly selected and analyzed using ImageJ 1.43j software (National Institutes of Health, Bethesda, MD).
Detection of RIG-I, ISG15, and B2M proteins
The cells were transduced as indicated in Figure 8. Western blots were performed using total cell lysate and anti-RIG-I/DDX58 (1:1000, ab238254; Abcam) or anti-ISG15 (1:1000, 2743S; Cell Signaling) primary antibodies were applied before detection using anti-goat (1:5000, A5420; Sigma) or anti-rabbit (1:5000, A0545; Sigma) secondary antibodies, HRP conjugated. Blots were visualized with ECL (GE Healthcare). Images were recorded using an ImageQuant LAS4000 imaging platform (GE Healthcare). Alternatively, the cells were harvested and submitted to flow cytometric detection of B2M. For this, the cells were incubated with the primary antibody specific for B2M (1:100, 316302; Biolegend, San Diego, CA), then the secondary antibody conjugated with Alexa Fluor-488 (1:500, 4408; Cell Signaling) before analysis using an Attune cytometer (Thermo Fisher Scientific).
Statistical analyses
Statistical analyses were performed using Prism software version 8.0 (GraphPad Software, San Diego, CA). Statistical significance was calculated as indicated in figure legends and were considered significant when p < 0.05. Each test was chosen because we propose that the data meet the assumptions of the test for the given situation. In vitro experiments were performed on at least three independent occasions using technical replicates as indicated in each figure legend. In vivo assays were performed with sample sizes as indicated in the figure legends, determined to permit analysis while respecting animal use guidelines.
Results
The luciferase-2 (Luc2), p14ARF and hIFNβ cDNAs were used in the construction of nonreplicating, RGD-modified Ad5 vectors where the constitutive CMV promoter is used to drive transgene expression (Supplementary Fig. S1). Transduction efficiency of the human melanoma cell lines with different TP53 gene status, UACC-62 (wt) and SK-Mel-29 (c.772G>A; pE258K) (Albino et al., 1994), was confirmed, revealing that an MOI of 100 was sufficient to transduce essentially 100% of cells of either lineage (Supplementary Fig. S2). The expression of each transgene, Luc2, hIFNβ, and p14ARF was confirmed by detection of luciferase activity, ELISA, and immunofluorescence microscopy, respectively (Supplementary Fig. S3A–C).
Transgene function was confirmed by qPCR detection of transcripts of key genes in the p53/ARF and IFNβ pathways (Fig. 1). As expected, IFNβ gene transfer resulted in increased transcription of genes associated with this pathway, including IRF1, 7, 9, as well as TRAIL in both cell lines. The regulation of p53-responsive genes, p21, HDM2, NOXA, and DNMT1, upon p14ARF gene transfer was clearly seen in UACC-62, as predicted by the presence of endogenous wild-type p53 in this cell line. Of note, when comparing p14ARF gene transfer to the nontransduced control group, increased expression of IRF7, IRF9, and TRAIL in the UACC-62 cell line was seen, providing the first indication that p14ARF may potentiate an anti-tumor immune response, especially in the presence of wild-type p53.
Before further examination of immune-related factors, the induction of oncolysis upon gene transfer was characterized. The impact of the single or combined p14ARF and hIFNβ gene transfer on cell proliferation and colony formation was assessed to reveal the short- and long-term cytotoxic effects. The growth curve (Fig. 2A) shows that the total number of viable cells was decreased for all conditions where transduction was performed. Although the control vector negatively impacted cell proliferation, p14ARF and hIFNβ, alone or in combination, further reduced viable cell counts for both the UACC-62 and SK-Mel-29 cell lines. For the colony formation assay, cells were transduced, collected, and re-seeded before observation of their capacity to adhere and continue proliferating, revealing that p14ARF alone or in combination with hIFNβ significantly inhibited colony formation (Fig. 2B).
Cell viability after gene transfer was assessed using an MTT assay and quantification of the hypodiploid (sub-G1) cell population was determined by flow cytometry after PI staining. The MTT assay shows that single gene transfer was effective for reducing mitochondrial activity relative to control vector treatment, especially for the UACC-62 cell line (Fig. 3A), yet combined gene transfer led to further reduction in mitochondrial activity compared with the single treatments for both cell lines. Note that data are normalized to the control vector, thus no Luc2 curve is presented in the graph. Accumulation of sub-G1 cells was induced by p14ARF alone or in combination with IFNβ gene transfer in UACC-62 (Fig. 3B), yet the combination of p14ARF and hIFNβ was superior to individual gene transfer for the induction of cell death in SK-Mel-29.
For each cell line, we examined cell death upon gene transfer in the presence or not of the pan caspase inhibitor Z-VAD-FMK (ZV). For UACC-62, gene transfer induces significantly more hypodiploid cells in the absence of ZV, suggesting that cell death is dependent on caspase activity. For SK-Mel-29, the accumulation of hypodiploid cells is not significantly different when ZV is or is not included in the treatment, indicating that caspase activity is not essential for cell death in this cell line. Thus, ZV has an effect in UACC-62, but not SK-Mel-29, suggesting that the mechanism of cell death differs between these two cell lines.
To further explore the possible mechanism of cell death, we evaluated three classic apoptosis events: activation of caspases 3 and 7, cleavage of PARP and exposure of phosphatidylserine on the cell surface. Although combined gene transfer activated caspase 3/7 activity in both cell lines, p14ARF was associated with a similarly high level of activity in UACC-62 (Fig. 4A). PARP was cleaved in all conditions where the p14ARF was delivered (Fig. 4B). Of interest, for UACC-62, annexinV/PI staining (Fig. 4C and Supplementary Fig. S4) was seen in the same conditions as caspase 3/7 activation and PARP cleavage. However, for SK-Mel-29, annexinV/PI staining was significant, although modest, upon combined gene transfer although this condition clearly activated caspase 3/7 and PARP cleavage. Taken together, these data show that p14ARF gene transfer alone or in combination with hIFNβ induces death of UACC-62 cells by a mechanism consistent with apoptosis. However, for SK-Mel-29, cell death upon combined gene delivery may involve additional mechanisms of cell killing that do not rely on caspase activity.
The assays of viability given in Figures 2 to 4 indicate that p14ARF is a driver of cell death, whereas hIFNβ reduces proliferation and metabolic activity in the cell lines studied here. Unexpectedly, the control vector encoding the Luc2 gene also reduced cell viability, MTT activity, and colony formation, but did not seem to influence the examination of hypodiploid cells, caspase 3/7 activity, or annexinV/PI staining.
In situ gene therapy was performed where s.c. UACC-62 tumors were formed in nude mice then treated with four doses intratumoral adenovirus injection. Combined gene transfer was superior for the inhibition of tumor progression as compared with single gene transfer (Fig. 5A). In parallel, tumors were recovered 48 h after the final treatment and subjected to immunohistochemical detection of Ki67. As given in Supplementary Figure S5, only combined gene transfer was associated with a decrease in tumor cell proliferation. Because tumor growth seemed to accelerate when treatments were ceased (day 7), we repeated this assay, comparing a four- and six-dose regimen, where two additional doses of combined adenovirus gene therapy were applied. In Figure 5B, the animals receiving a total of six doses showed prolonged survival as compared with those receiving four rounds of treatment. Thus, tumor response to treatment was dose dependent. In vivo experimentation with SK-Mel-29 was not possible because these cells did not readily form tumors in nude mice.
We next explored the potential of p14ARF and hIFNβ gene transfer to trigger ICD by assessing the emission of DAMPs, such as calreticulin, ATP, and HMGB1. Strikingly, significant detection of calreticulin at the cell surface was most clearly seen when cells were treated with combined gene transfer (Fig. 6A). The secretion of ATP (Fig. 6B) was induced by p14ARF alone or in combination with IFNβ. In contrast, HMGB1 was clearly released upon p14ARF treatment in UACC-62 and was detected in SK-Mel-29, especially upon combined gene transfer (Fig. 6C). These assays suggest that p14ARF was sufficient to induce the emission of three critical ICD markers in the UACC-62 cell line, whereas SK-Mel-29 required combined gene transfer.
To further explore the role of p14ARF in as an immune stimulator, we used qPCR to evaluate the expression of a variety of factors that may potentiate immune activation. As given in Figure 7, p14ARF gene transfer activated the transcription of pattern recognition receptors TLR3, DDX58 (which encodes RIG-I), and IFIH1, as well as ISG15 (part of the interferon pathway), and factors related to antigen processing and presentation, TAP-1 and B2M, especially in the UACC-62 cell line. In general, the use of p14ARF gene transfer was more effective in altering gene expression than the use of Nutlin-3, a pharmacologic antagonist of MDM2. As expected, IFNβ gene transfer was sufficient to regulate the transcription of these genes (Supplementary Fig. S6). At the protein level, increased RIG-I and ISG15 expression was confirmed for both cell lines by western blot while elevated B2M, examined by flow cytometry, was seen only for the UACC-62 cell line (Fig. 8 and Supplementary Fig. S7).
Additional genes examined (Supplementary Fig. S8) reveal a role for IFNβ alone or in combination with p14ARF for the regulation of endogenous retroviruses, pattern recognition receptors, and antigen processing. P14ARF gene transfer regulates several factors predicted to potentiate an anti-tumor immune response, especially in the presence of wild-type p53, as is the case for the UACC-62 cell line. Overall, we encountered differential expression more often for the UACC-62 (wild-type p53) cell line than for SK-Mel-29 (mutant p53) when p14ARF gene transfer was performed, likely owing to the status of endogenous p53. On the contrary, the response to IFNβ gene transfer was similar between the cell lines tested in this study.
Discussion
In this work we tested our p14ARF and hIFNβ gene therapy approach in human melanoma cell lines that differ in their p53 status to reveal oncolysis and predictors of immune activation. Strikingly, an unexpected role was exposed for p14ARF in the induction of factors that may potentiate an anti-tumor immune response. In UACC-62, p14ARF gene transfer was sufficient to induce oncolysis, cell death by a mechanism consistent with apoptosis, ICD, and upregulate the expression of several genes related to immune activation. For SK-MEL-29, combined gene transfer was more effective for the induction of cell death and induction of ICD markers. We do not expect that endogenous p53 status alone will be sufficient to predict response to gene transfer, but we now have evidence that p14ARF plus IFNβ may be effective even in p53-mutant cell lines.
In our previous work we focused on cell lines that retained p53 in the wild-type form, a typical situation seen in melanoma (Giglia-Mari and Sarasin, 2003), and applied a vector that uses a p53-dependent promoter to control transgene expression (Merkel et al., 2013; Medrano et al., 2016; Hunger et al., 2017; Cerqueira et al., 2020; David et al., 2020; Del Valle et al., 2021). However, the use of a p53-responsive promoter may be confounded in p53-deficient cells. For this reason, we constructed new vectors using the constitutive CMV promoter, allowing us to explore a variety of cell lines, including those that harbor mutant p53.
Throughout our studies we have been careful to examine both human and mouse cell lines because they differ in their response to IFNβ (Qin et al., 2001). Here and in our recent studies, we have focused on gene transfer to human cells using vectors encoding human cDNAs, whereas our previous studies have focused on the transfer of mouse cDNA to mouse cells (Strauss et al., 2018). Different from our prior observations in mouse melanoma (Hunger et al., 2017), here p14ARF seems to be quite effective for the induction of cell death, perhaps more so than hIFNβ, when each is delivered alone.
The induction of cell death accompanied by the emission of immunogenic factors is typical of virotherapies where virus replication and/or induction of an antiviral response induces oncolysis (Galluzzi et al., 2020; Lemos de Matos et al., 2020). We consider our combined gene transfer approach to be an inducer of oncolysis although we employ a nonreplicating vector.
One possible advantage of our approach is that, without viral replication, we are free to express an antiviral cytokine, IFNβ, as part of our strategy. Many oncolytic vectors rely, in part, on the lack of an effective type I interferon response in tumor cells, making them permissive for virus replication, whereas normal, healthy cells maintain the capacity for interferon signaling and thus restrict virus replication (Allagui et al., 2017; Ebrahimi et al., 2017; Fend et al., 2017; Ying et al., 2017; Kurokawa et al., 2018; Lee et al., 2019; Geoffroy and Bourgeois-Daigneault, 2020). However, innate and adaptive immune activation involves type I interferon for a variety of functions, including activation of dendritic cells and recruitment of T cells (Medrano et al., 2017). Thus, the lack of interferon response is beneficial to the oncolytic virus because it escapes potential antiviral defenses, but detrimental to anti-tumor immune activation. Induction of oncolysis while supporting the type I interferon response would be expected to capitalize on both activities. As such, the proposal of using a replicating vector to induce cell death and also the need to restore IFN signaling to support the anti-tumor immune response may be at odds, although such approaches have been reported (Kurisetty et al., 2014; Geoffroy and Bourgeois-Daigneault, 2020). Because our vectors do not replicate, we can include IFNβ as a therapeutic transgene without concern that it will inhibit viral activity.
Alternatively, as seen in the UACC-62 cell line, the transfer of p14ARF resulted in the activation of several mediators of the interferon pathway as well as emission of ICD markers. This suggests that at least for some cells, p14ARF gene transfer may contribute to an anti-tumor immune response. This finding is consistent with the observations of Zhou et al. (2021) who used pharmacological inactivation of MDM2/MDMX to liberate p53 and, in turn, promote viral mimicry and an anti-tumor immune response. In fact, we have previously shown that, in combination, p19Arf, IFNβ, and the adenoviral vector itself all contribute to an antiviral response that we consider to be an important component of our approach (Hunger et al., 2017).
In this study, for the first time, we show that p14ARF gene transfer can also elicit cellular responses that are expected to promote an anti-tumor immune response, although the transfer of p14ARF to a p53-mutant cell line (SK-Mel-29) was less effective for the upregulation of immune activating genes and the combined gene transfer of p14ARF and IFNβ may be better suited. Further assays would be required to fully understand the role of p53, thus we cannot assert that p53 status will be predictive of cellular response.
The in situ gene therapy assays showed a subtle, but clear, advantage when the combination of p14ARF and hIFNβ were applied to the s.c. UACC-62 tumors. Because the two additional rounds of treatment were associated with prolonged survival, it is possible that the treatment regimen may be further optimized, although this was not explored here. These in vivo assays were met with some technical issues. The s.c. UACC-62 tumors, whether treated or not, were highly hemorrhagic, a situation that may have influenced the measurement of tumor volume. Our vectors encode the luciferase-2 transgene, included with the intention of monitoring the extent of virus expression in the treated tumor. Although this was not performed, it would not have revealed tumor progression, only presence of cells expressing the vector. In addition, the SK-Mel-29 cells did not readily form s.c. tumors as attempted here, thus we were unable to explore the treatment of these cells in vivo. We did not have the opportunity to explore other conditions that may have favored SK-Mel-29 tumor formation, such as coinjection with components of the extracellular matrix and/or increasing the number of cells, as has been reported (Emmrich et al., 2009).
We are encouraged by the results shown here and postulate that in an immunocompetent model, the ICD and immune-stimulatory factors induced by our combined gene transfer approach may contribute to greater control over tumor progression owing to the expected anti-tumor immune response, a point we have explored previously (Catani et al., 2016; Medrano et al., 2016).
With this study we have expanded our understanding of the combined p14ARF and IFNβ gene transfer approach. Although much work lies ahead, we are encouraged by the new pro-immunogenic role of p14ARF, including its induction of oncolysis accompanied by the emission of ICD markers and upregulation of genes involved in immune regulation, especially in the presence of wild-type p53.
Footnotes
Acknowledgments
The authors thank Roger Chammas (ICESP-FMUSP) for unwavering support, Mara Junqueira (Centro de Medicina Nuclear-FMUSP) for assistance with the in vivo assays and Allane dos Santos Ferreira Maranhão (ICESP-FMUSP) for help with tissue processing.
Authors' Contributions
S.A.M., F.A.: designed and performed assays, elaborated and edited text and figures, interpreted results; O.L.D.C., P.R.D.V., A.H., P.V.S.O., B.B.: performed assays and edited text; E.C.S.: designed assays and interpreted results; B.E.S.: designed assays, interpreted results, elaborated and edited text, secured funding.
Disclosure Statement
The authors state that they have no conflicts of interest regarding the work presented here.
Funding Information
This work was supported by the Sao Paulo Research foundation (FAPESP) grant 2015/26580-9 (B.E.S.) and fellowships 2018/04800-5 (F.A.), 2017/23068-0 (O.L.D.C.), 2013/16074-3 (P.R.D.V.), 11/10656–5 (A.H.). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001 (S.A.M.). Funding was also provided by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), fellowship 302888/2017/9 (B.E.S.).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.