
Abstract
Endoplasmic reticulum (ER) stress and oxidative stress (OS) are often related states in cells as part of normal physiology but more frequently manifested in the pathophysiology of many diseases, particularly diseases involving acute or chronic inflammation. In this study, we reviewed recent findings about the role of ER stress and OS in the pathogenesis of inflammatory diseases.
Introduction to the Endoplasmic Reticulum Stress and Oxidative Stress
The endoplasmic reticulum (ER) is the largest organelle of eukaryotic cells, comprising the rough ER constituted by sheets, and the smooth ER constituted by tubules, each structure is related to the type of processes that takes place at the site (Oakes and Papa, 2015). ER performs various cellular functions such as protein folding, post-translational modifications, fatty acid and sterol biosynthesis, xenobiotic detoxification, and intracellular calcium storage (Riaz et al, 2020). When ER encounters a substantial increase in workload, some proteins cannot be folded properly and natively leading to aggregation and accumulation of the unfolded proteins in the ER lumen. Such a scenario is known as ER stress (Burman et al, 2018). Severe and prolonged ER stress is thought to drive the pathology of many chronic disorders owing to its potential to elicit aberrant inflammatory signaling and facilitate cell death.
In response to ER stress, cells initiate the unfolded protein response (UPR) that involves a series of signaling and transcriptional events in an attempt to restore ER homeostasis by ways of increasing protein folding capacity, diminishing translation rate, and degrading unfolded and misfolded proteins (Chen and Cubillos-Ruiz, 2021). The canonical UPR signaling is triggered by activation of three ER membrane-bound transducers: (1) the endoribonucleases inositol requiring enzyme 1 (IRE1α) (ubiquitously expressed) and IRE1β (confined to mucosal secretory cells, which splice the X-box-binding protein (XBP1) mRNA leading to translation of the sXBP1 transcription factor that drives expression of chaperones and other ER-resident proteins required for folding; (2) the ER-resident protein kinase RNA-like ER kinase (PERK) that inhibits translation through eIF2a and induces transcriptional responses by activating transcription factor-4 (ATF4) and CCAAT/enhancer-binding protein homologous protein (CHOP) resulting in ER stress–induced apoptosis; and (3) the activating transcription factor 6 (ATF6) that also facilitates the generation of proteins for enhancing ER function (Chen and Cubillos-Ruiz, 2021).
Oxidative stress (OS) is a form of cellular stress and damage caused by excess production and accumulation of reactive oxygen species (ROS) that compromises antioxidant defense mechanisms (Ren et al, 2021). ROS are a group of small reactive molecules which could bring some beneficial effects on cellular responses (Kattoor et al, 2017). For an instance, accumulating evidence has suggested that intracellular ROS would act as secondary messengers in intracellular signaling cascades, which induce and maintain physiological responses to protect cells from hypoxia and inflammation (Zepeda et al, 2013). However, abnormal production and accumulation of ROS have detrimental effects on cellular function and homeostasis because it could trigger many cellular signaling pathways and induce damage to DNA, proteins, and cells (Henriksen et al, 2011, Tsai et al, 2014). Persistent exposure to ROS upregulates the production of oxidatively modified nucleic acids, lipids, and proteins leading to the development of diseases (Srinivas et al, 2019).
Crosstalk Between ER Stress and OS
Although the association between ER stress and OS remains to be fully elucidated, studies have supported the notion that these two cellular stresses are closely linked in many physiological and pathological conditions (Victor et al, 2021). In ER, redox homeostasis is critical for the protein folding process and disulfide bond formation (Maamoun et al, 2019). Alterations in ER-mediated protein folding pathways can cause an ROS imbalance and enhance ROS production, indirectly disturbing both ER and redox homeostasis (Hasanain et al, 2015). In an ER stress condition, misfolded proteins particularly with aberrant disulfide bond formation and reduction may cause ROS accumulation and diffusion into the cytoplasm leading to cellular OS (Margittai et al, 2012; Mennerich et al, 2019). Conversely, OS from increased production of ROS can induce protein misfolding and ER stress as several oxidants including peroxides, metal ions, and oxidation byproducts can pathologically initiate the UPR (Cao and Kaufman, 2014). It appears that ER stress and OS are intrinsically linked with one process triggering the other in various scenarios encountered by the cell.
ER Stress and OS in Inflammatory Cytokine Production
It has been established that ER stress response is one of the major factors causing inflammatory pathologies through regulation of production of the proinflammatory cytokines. Inflammation is usually triggered when pattern recognition receptors, such as Toll-like receptors (TLRs) or nucleotide-binding oligomeric domain (NOD)-like receptors (NLRs), detect tissue damage or microbial infection. Kevin Shenderov et al (2014) showed that ER stress can promote the secretion of mature interleukin (IL)-1β from macrophages upon TLR4 stimulation through a caspase-8- and TRIF-dependent pathway. Treatment of macrophages with ER stress–inducing reagents tunicamycin or thapsigargin followed by TLR stimulation–induced IL-1β and TNF-α production (Komura et al, 2013). Intriguingly, ER stress has recently been reported to associate with inflammasomes in inflammatory signaling pathways (Chen et al, 2019; Lebeaupin et al, 2015).
The inflammasome is a signaling protein complex composed of NLR including caspase 1 and NLR family pyrin domain containing 3 (NLRP3) and takes a crucial part in different inflammation-induced autoimmune and metabolic disorders (de Torre-Minguela et al, 2017; Gong et al, 2017; Perera et al, 2017; Ralston et al, 2017). Upon activation, inflammasome can convert procaspase 1 into active caspase 1 that subsequently activates pro-IL1β into active IL-1β, one of the key mediators of the inflammatory response. The hypothesis that ER stress is strongly associated with inflammasome during inflammation has now been clarified from several studies demonstrating that ER stress is involved in the activation of pro-IL1β and NLRP3 (Lerner et al, 2012; Menu et al, 2012) (Fig. 1).
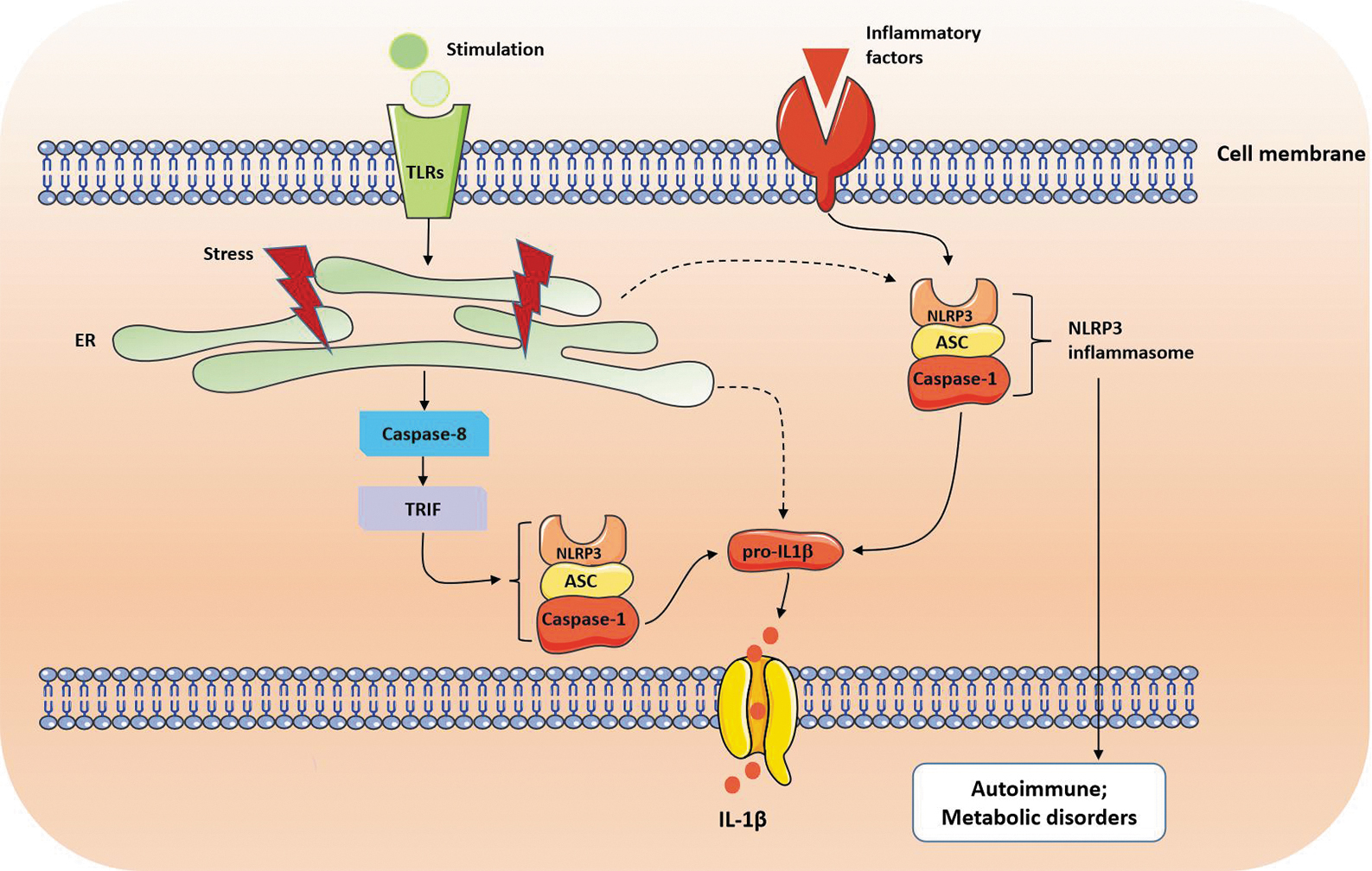
Crosstalk between ER stress, NLRP3 inflammasome, and inflammation. ASC, apoptosis-associated speck-like protein containing a caspase-1 recruitment domain; ER, endoplasmic reticulum; IL-1β, interleukin-1β; NLRP3, NLR family pyrin domain-containing protein 3; Pro-IL1β, pro-interleukin-1β; TLRs, Toll-like receptors; TRIF, Toll/IL-1R domain-containing adapter-inducing IFN-β.
In parallel with ER stress, when the redox equilibrium is disturbed owing to the excessive accumulation or depletion of ROS, many cellular signaling pathways are influenced, which confers to the cellular dysfunction and subsequently the development of various pathologies. ROS act as second messengers and activate numerous signal transduction pathways including PI3K/Akt, p38 MAPK, ERK, JNK, PKC, Src family kinases, and growth factor tyrosine kinase receptor pathways (Zhang et al, 2016), all of which could lead to the production of proinflammatory cytokines.
Moreover, ROS could also stimulate redox-sensitive transcription factors such as NF-κB, NRF2, and hypoxia-inducible factor (HIF)-1 that all play important roles in inflammatory responses (Lee and Yang, 2012). NF-κB transcription factor, which is critical for a wide range of processes, such as immunity, inflammation, cell development, growth, and survival, can also regulate ROS production and degradation (Wang et al, 2019b). NRF2 is a transcription factor that primarily regulates the antioxidant protein expression in response to oxidative damage can lead to inflammatory signaling as well (Kudo et al, 2020; Nishimoto et al, 2017). Under the normoxic condition, NRF2 is retained in the cytoplasm and degraded by Kelch-like ECH-associated protein 1 (KEAP1) and Cullin3 protein to prevent the activation of associated downstream signaling pathways (Guo et al, 2022). During oxidative damage, KEAP1-Cullin3-induced NRF2 degradation is inhibited as such NRF2 translocates to the nucleus where it binds to the antioxidant response element promoter to upregulate the gene expressions for antioxidant proteins (Cullinan and Diehl, 2006). It has also been shown that knockout of HIF-1α in the myeloid cells increases the survival rates in a murine LPS-induced sepsis model and decreases serum levels of proinflammatory cytokines TNF-α, IL-1α, IL-1β, and IL-12 (Peyssonnaux et al, 2007) (Fig. 2).
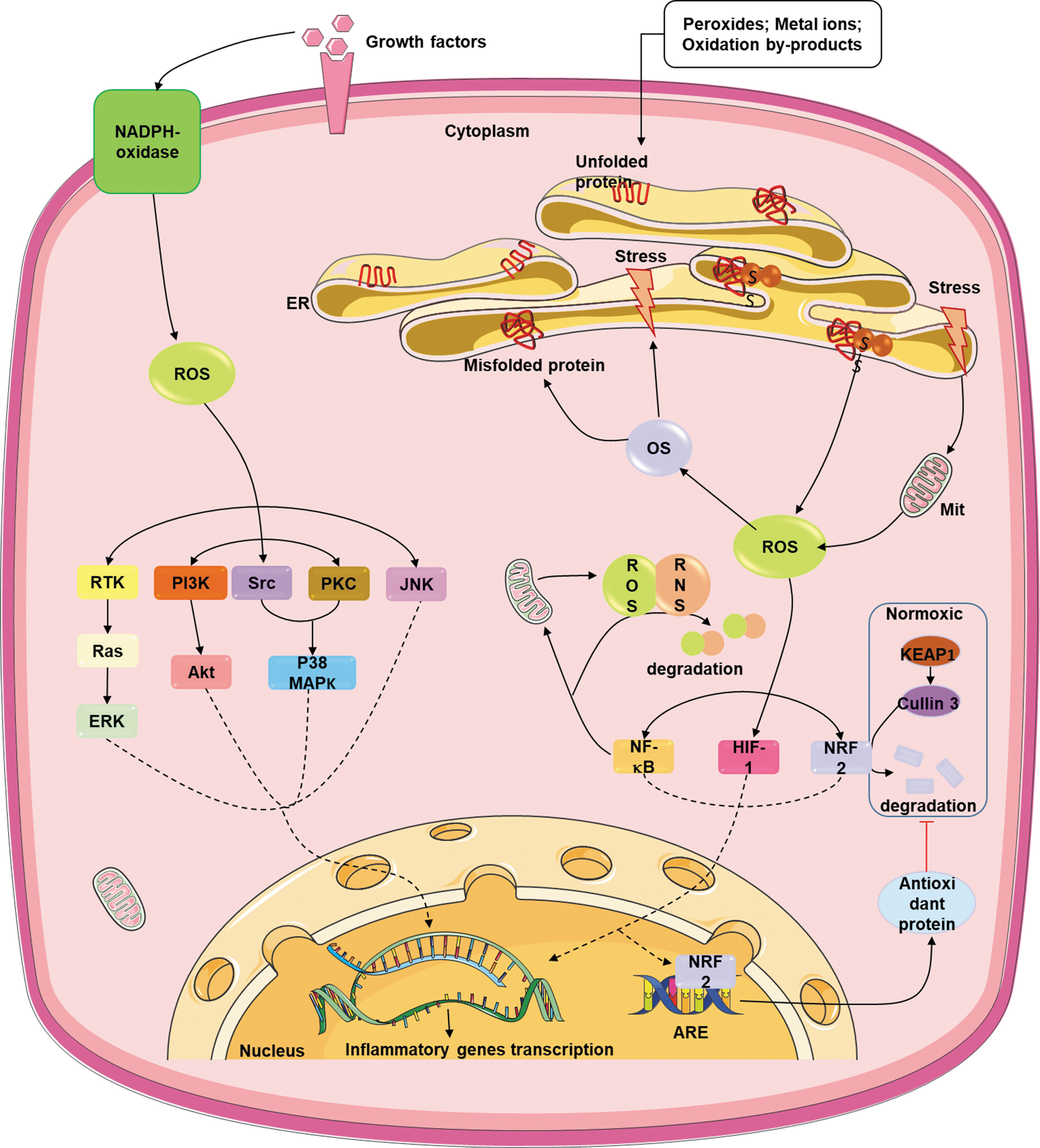
Crosstalk between ER stress, OS, and inflammatory cytokine production. Akt, protein kinase B; ARE, antioxidant response element; ERK, extracellular regulated protein kinases; HIF-1, hypoxia-inducible factor 1; JNK, c-Jun N-terminal kinase; KEAP1, Kelch-like ECH-associated protein 1; MAPK, mitogen-activated protein kinases; Mit, mitochondria; NF-κB, nuclear factor-kappa B; NRF2, nuclear factor erythroid-2-related factor (NF-E2-related factor 2); OS, oxidative stress; PI3K, phosphatidylinositol 3-kinase; PKC, protein kinase C; PTK, receptor tyrosine kinase; Ras, rat sarcoma protein; RNS, reactive nitrogen species; ROS, reactive oxygen species; SS, disulfide bond; Src, src protein.
ER Stress and OS in Inflammatory Diseases
ER stress and OS have been shown to contribute to the pathogenesis of inflammatory disorders in various tissue or organ systems such as inflammatory bowel disease (IBD), chronic obstructive pulmonary disease (COPD), chronic kidney disease, viral hepatitis, pancreatitis, rheumatoid arthritis (RA), and others (He et al, 2018; Laudisi et al, 2019; Lee et al, 2018; Liong and Lappas, 2016; Wang et al, 2019a; Zha et al, 2015). Their respective targets and mechanisms have been described in many reviews. This review briefly summarizes the target organs/tissues of ER stress and the target genes and effects of activation of hypoxia-inducible factor (HIF)-1, an important molecule involved in OS. The following part mainly focuses on their crosstalk in a variety of inflammatory diseases (Tables 1 and 2).
Target Organs/Tissues of Endoplasmic Reticulum Stress and Inflammation Crosstalk
ATF6, activating transcription factor 6; eIF2α, eukaryotic translation initiation factor 2α; ER, endoplasmic reticulum; HMCs, human glomerular mesangial cells; IRE1, inositol-requiring enzyme 1α; JNK, c-Jun NH2-terminal kinase; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor-kappa B; OS, oxidative stress; PERK, pancreatic endoplasmic reticulum kinase; RANKL, nuclear factor-kappa B ligand; TLR, Toll-like receptor; UPR, unfolded protein response; XBP1, X-box protein 1.
Target Genes and Effects of Activation of Hypoxia-Inducible Factor-1 and Inflammatory Response
ABCA1, ATP-binding cassette transporters A1; COX4-2, cytochrome c oxidase4–2; EPO, erythropoietin; HIF-1, hypoxia-inducible factor 1; IGF2, insulin-like growth factor 2; IL, interleukin; iNOS, inducible nitric oxide synthase; LEP, leptin; MCP-1, monocyte chemoattractant protein-1; MIF, macrophage migration inhibitory factor; MMP-1, MMP-2, MMP-3, matrix metalloproteinases 1–3; NO, nitric oxide; PDGF, platelet-derived growth factors; PDGFR, platelet-derived growth factor (PDGF)/PDGFR receptor; PHD2, prolyl hydroxylase domain protein 2; PTPs, protein tyrosine phosphatases; SDF-1α, stromal cell–derived factor-1α; THBS1, thrombospondin 1; VEGF, vascular endothelial–derived growth factor.
Inflammatory bowel disease
The intestinal epithelium is constantly exposed to a microbial environment and is highly susceptible to extracellular and intracellular challenges (Kaser et al, 2013). IBD, including Crohn's disease and ulcerative colitis, is a group of inflammatory pathologies in the gastrointestinal tract. Ample evidence indicates that intestinal epithelial cells (IECs), particularly Paneth and goblet cells, are susceptible to changes in ER protein folding homeostasis as a consequence of environmental challenge and/or genetic defects (Cao et al, 2013; Gagliardi et al, 2021; McGuckin et al, 2011). Both in the mucosal tissues of patients with IBD and several murine models of colitis and Crohn's ileitis, ER stress markers are frequently identified (Engevik et al, 2021; Rees et al, 2020; Xu et al, 2020). Mice expressing a misfolding-prone mutant of MUC2 mucin, the major mucin in the large intestine, are manifested with ER stress, goblet cell dysfunction, and spontaneous colitis (Heazlewood et al, 2008).
Of interest, glucocorticoids, a family of drugs used in clinics for the treatment of IBD for decades, were found to attenuate ER stress in colonic epithelial cells via a mechanism of transactivating ER chaperones and ERAD components (Das et al, 2013). In animal models, the deletion of the UPR component XBP1 impairs the ability of intestinal epithelium to resolve ER stress ultimately leading to spontaneous intestinal inflammatory disorder with characteristics of human IBD (Kaser et al, 2013; Kaser et al, 2008). In fact, XBP1 malfunction is regarded as a genetic risk factor for both Crohn's disease and ulcerative colitis (Glas et al, 2012; Kaser et al, 2008). Although the precise molecular mechanism of how ER stress and the UPR regulate inflammation in IECs and inflammatory cells during IBD development and progression is yet to be elucidated, those prior studies support the overall conclusion that ER stress–induced epithelial dysfunction may be sufficient enough to inflict intestinal inflammation through impairment of mucosal homeostasis and barrier function in the gut.
It has been demonstrated that ROS is increased in the mucosal tissues of both chemical-induced and genetic models of IBD as well as in patients with IBD (Aviello and Knaus, 2017; Wendland et al, 2001). In the course of IBD initiation and progression, multiple cell types in the gut such as neutrophils, macrophages, and IEC can produce a large amount of superoxide. Overproduction of ROS, such as superoxide anion, which results in hydrogen peroxide and oxygen, contributes to intestinal inflammation by inducing epithelial cell death and mucosal tissue injury as well as igniting cell-autonomous inflammation in both IEC and inflammatory cells (Balmus et al, 2016; Zhu and Li, 2012). The depletion of antioxidants was also observed in inflamed mucosal tissues in both human and animal models (Esworthy et al, 2001; Khor et al, 2006). The concept that endogenous antioxidative stress defense plays an important role in the homeostasis of the intestinal mucosa was supported by studies of mice deficient in glutathione peroxidase-1/2 or Nrf2 (Esworthy et al, 2001; Khor et al, 2006).
It should be realized that although ER stress and OS may coexist in inflammatory mucosa, it is still unclear whether the two cellular stresses reciprocally induce each other in the gut or whether either one is adequate to initiate and promote IBD.
Infectious respiratory disease
ER stress and OS have been reported to participate in the pathophysiology of several nonmalignant diseases of lung including infections, cystic fibrosis, idiopathic pulmonary fibrosis, and asthma (Chen et al, 2018). In the case of infections, ER and OS could arise from direct effects of the pathogen on infected cells or as a consequence of the immune response to the pathogen. For instance, infection of respiratory epithelial cells with influenza A virus activates the UPR, which is driven by IRE1, but against the backdrop of PERK and ATF6 suppression (Hassan et al, 2012). As with chronic inflammatory lung diseases (such as COPD and chronic bronchitis), another mechanism by which infection leads to respiratory oxidation and ER stress is through the activation of immune system mediators such as cytokines and ROS (mainly hydrogen peroxide) (Barreiro et al, 2019; Wiegman et al, 2020). COPD and chronic bronchitis are characteristic of OS, inflammation, and mucus hypersecretion, all of which could drive ER stress under these circumstances (Tawiah et al, 2018).
Chronic kidney disease
The mechanism underlying nephrotoxicity by patulin (PAT), a metabolite produced by several species of the genera of Penicillium, Aspergillus, and Byssochlamys, has been linked to the induction of ER stress and production of excessive ROS, such as superoxide anion and hydrogen peroxide (Boussabbeh et al, 2016). Consistently, the two common dietary compounds quercetin (a natural flavonoid) and crocin (a natural carotenoid) could prevent PAT-induced apoptosis by inhibiting the ROS-mediated ER stress pathway. Some drugs with severe nephrotoxic side effects such as acetaminophen cause renal tubular injury via ROS-mediated ER stress as evidenced by PERK pathway activation, CHOP induction, and caspase-12 cleavage (Lorz et al, 2004; Ściskalska et al, 2015).
ER stress is also involved in age-related renal fibrosis. It has been reported that the age-linked aggregation of oxidative carbonylated Bip/Grp78 or PDI could lead to ER stress–induced kidney dysfunction (Naidoo, 2009). Likewise, the ER marker Bip/Grp78 was highly expressed in kidney biopsy specimens of patients with uromodulin-linked renal disease (Adam et al, 2012). These findings implicate that both ER stress and OS are key mediators for chemical-induced renal disease and that these two cellular stresses may also serve as protective mechanisms against kidney injury.
Viral hepatitis
ER stress–mediated ROS, free radicals, and superoxide anion are involved in the pathogenesis of different liver diseases including acute and chronic viral hepatitis (Apostolova et al, 2013; Duvigneau et al, 2019; Ríos-Ocampo et al, 2019). It has been shown that HBV and HCV replication and expression of viral proteins could induce cellular stress contributing to the pathogenesis of liver injury and liver fibrogenesis (He et al, 2018; Ke and Chen, 2012). The alterations in cellular homeostasis owing to the infection can lead to an increase of OS and/or ER stress. Increased OS has been found in liver biopsies from patients with chronic HCV infection and the levels of superoxide radicals and hydrogen peroxide were significantly higher in patients infected with HCV compared with other liver diseases (Ivanov et al, 2013; Valgimigli et al, 2002).
Furthermore, the free radical–mediated lipid peroxidation, steatosis, and prooxidant markers were also elevated during chronic HCV infection (Farinati et al, 1995). In acute hepatic disease, the GSH level is significantly decreased and the accumulation of peroxide accelerates protein aggregation and ER stress, which may increase the development of disease and reduce hepatic functional efficiency for chronic progression (Galligan et al, 2012).
In addition, studies have confirmed that ER stress induction by HBV infection has been implicated in liver carcinogenesis and disease progression with chronic inflammation through enhanced inflammation and OS-mediated DNA damage (Choi et al, 2019; Lorz et al, 2004).
Pancreatitis
Acinar cells in the pancreas are specialized in the production, storage, and secretion of various digestive enzymes and other proteins. To ensure this high rate of protein production, acinar cells are evolved to possess extensive ER networks. The role of ER stress responses has been extensively investigated in alcohol-related pancreatitis (Lugea et al, 2011). It is apparent that a functional UPR effectively alleviates ethanol-induced ER stress and protects from ethanol-induced pancreatic damage because partial deletion of XBP1, the essential component of UPR significantly increased pancreatic damage in the XBP1+/− mice fed with ethanol (Lugea et al, 2011). The role of the functional ER responses in other forms of pancreatic injury is currently uncertain (Sah and Saluja, 2011).
Although OS is generally regarded as a detrimental factor in pancreatitis, limited success is achieved in trials using antioxidant agents (Mohseni Salehi Monfared et al, 2009; Sateesh et al, 2009). A study investigating the role of ROS (peroxidation products) in pancreatitis yielded unexpected results (Booth et al, 2011). In this study, peroxide induction in the acinar cells by bile acid promoted apoptosis, whereas inhibition of peroxide generation led to necrosis accompanied by a significant reduction in ATP production, indicating that ROS generation within acinar cells functions as a protective response during pancreatitis. Meanwhile, OS in the neutrophils that were activated in response to acinar injury may be responsible for further dissemination of local and systemic inflammation (Booth et al, 2011). Thus, OS itself appears to have a dual role in the pancreatic injury.
Rheumatoid arthritis
In the synovial fluid of patients with RA, it has been found that the IRE1α/XBP1-mediated UPR branch is activated in macrophages (Qiu et al, 2013). In the animal model of inflammatory RA, IRE1α facilitates the production of proinflammatory cytokines from macrophages including IL-6, TNF-α, IL-1β, and RANTES. In macrophages and neutrophils, TLR interacts with IRE1α via signaling through MyD88 and TNF receptor–associated factor 6 (TRAF6) to activate IRE1α, thus promoting the expression of the genes encoding proinflammatory cytokines (Qiu et al, 2013). TLR-mediated IRE1α activation by TRAF6 acted through catalyzing IRE1α ubiquitination and blocking the recruitment of protein phosphatase 2A (PP2A), a phosphatase known to dephosphorylate and inactivate IRE1α.
OS also executes an essential part in the pathophysiology of RA (Phull et al, 2018). The imbalanced antioxidant system and higher lipid peroxidation levels in serum and synovial fluid have been reported in RA (Mateen et al, 2016). Although it is an indirect confirmation, the role of ROS in ligament degradation is evidenced from the presence of peroxidation products in the cartilage, and modified low-density lipoprotein (LDL), a nitrous type II collagen peptide, and oxidized IgG in the serum and urine of the RA patients (Hitchon and El-Gabalawy, 2004; Phull et al, 2018). Based on the observation that nitrated proteins, nitrotyrosine, and oxidized LDL were aggregated in the cartilage of patients suffering from arthritis, an immediate complication of peroxidation products in chronic arthritis has been proposed (Ahmed et al, 2016; Henrotin et al, 2003). Moreover, it was demonstrated that a low antioxidant status appeared to be a risk factor for initiating and developing RA in a Finnish cohort of 1419 adult men and women (Heliövaara et al, 1994) (Fig. 3).
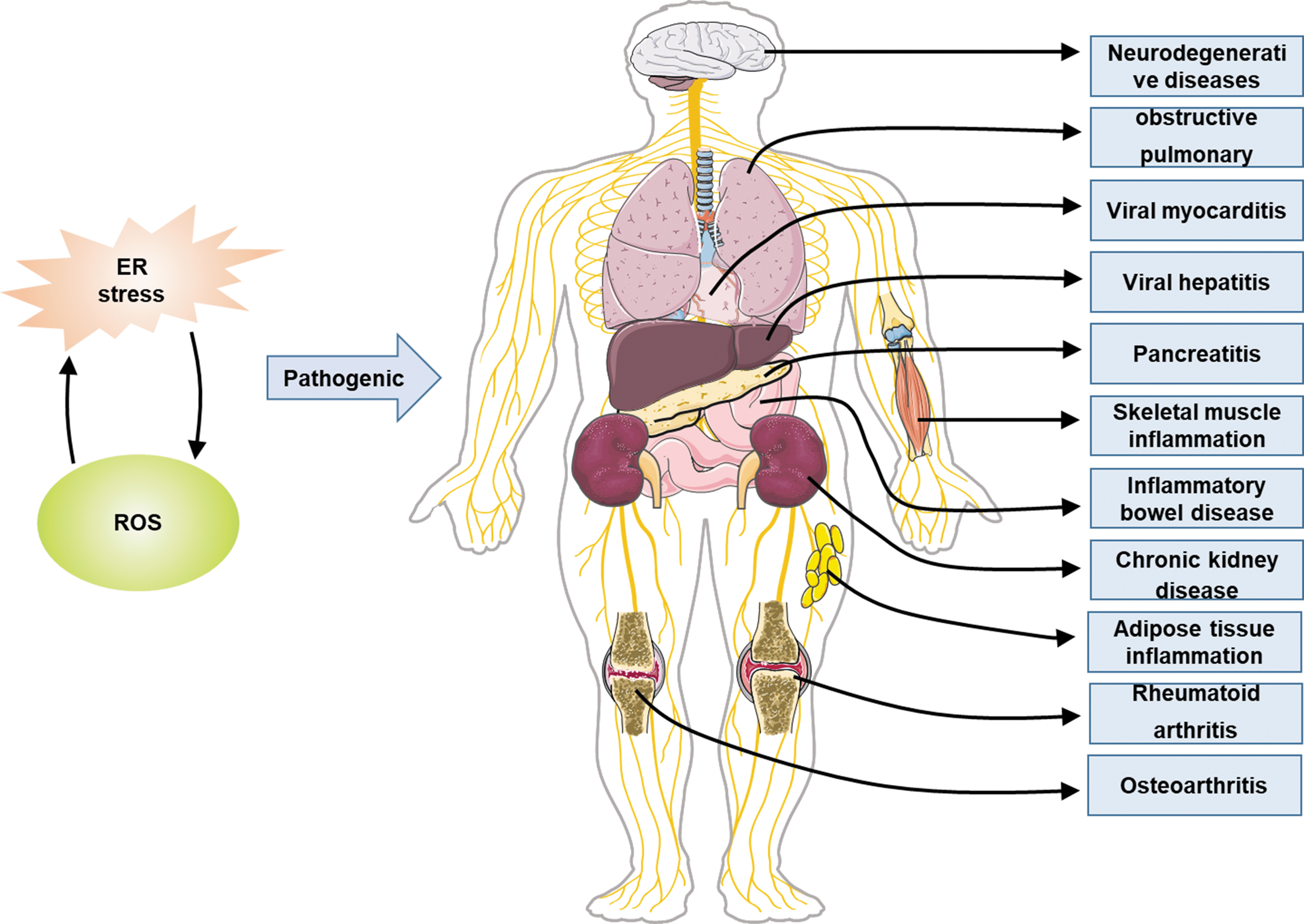
ER stress and OS related to inflammatory diseases.
Conclusions and Perspectives
Over the past few decades, we have witnessed hot and fast advancing research topics on the ER stress response, OS, and inflammation as well as their interactive relationships in the biomedical field. However, an in-depth mechanistic understanding of the crosstalk or relationship between the intracellular stresses and inflammatory responses and their participation in disease progression has not yet been fully accomplished. Future studies are needed to understand the pathophysiology behind cellular stress-mediated alterations in the protein folding and misfolding processes and to dissect the precise mechanism(s) of the interactions between ER stress and OS signaling in the context of inflammation with the hope of making them lucrative targets for therapeutic benefit to curing many dreadful diseases related to inflammation.
Footnotes
Acknowledgment
The authors thank everyone for their help with this review.
Authors' Contributions
All the authors have approved to publish this article. Y.T. and X.Z. wrote the article. T.C., E.C., Y.L., W.L., Y.H., and B.H. revised the article. S.L. critically revised the article for important intellectual content. All authors read and approved the final version of the article.
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Natural Science Foundation of China (No. 81201331), the Natural Science Foundation of Hunan Province (2022JJ30532) and Hunan Provincial Innovation University of South China Innovation Foundation for Postgraduate (No. 213YXC016).