
Abstract
Cell adhesion and stable signaling regulation are fundamental ways of maintaining homeostasis. Among them, the Wnt/β-CATENIN signaling plays a key role in embryonic development and maintenance of body dynamic homeostasis. At the same time, the key signaling molecule β-CATENIN in the Wnt signaling can also function as a cytoskeletal linker protein to regulate tissue barriers, cell migration, and morphogenesis. Dysregulation of the balance between Wnt signaling and adherens junctions can lead to disease. How β-CATENIN maintains the independence of these two functions, or mediates the interaction and balance of these two functions, has been explored and debated for a long time. In this study, we will focus on five aspects of β-CATENIN chaperone molecules, phosphorylation of β-CATENIN and related proteins, epithelial mesenchymal transition, β-CATENIN homolog protein γ-CATENIN and disease, thus deepening the understanding of the Wnt/β-CATENIN signaling and the homeostasis between cell adhesion and further addressing related disease problems.
Introduction
β-CATENIN is a multifunctional protein that is well conserved during evolution and development from protists to animals (Valenta et al, 2012). It functions in a dual role in cells to control embryonic patterning and organogenesis (Brembeck et al, 2006). It is not only the most important nuclear effector of the Wnt/β-CATENIN signaling, but also as a cytoskeletal junction protein that maintains cell adhesion and is critical for cadherin-based adherens junctions. Implementation of the dual function of β-CATENIN is based on the two functional pools of β-CATENIN, the transcriptional pool, and the adhesive pool (van der Wal and van Amerongen, 2020). How β-CATENIN maintains the independence of these two pools, or the interaction and balance between them, has long been explored and debated.
In the transcriptional pool, β-CATENIN is the core of the Wnt/β-CATENIN signaling. When Wnt ligands (Wnts) bind to the frizzled/lipoprotein receptor-related proteins 5 and 6 (Lrp5/6), signals are generated and transmitted downstream. The destruction complex formed by glycogen synthase kinase 3-beta (GSK3β), axin, and adenomatous polyposis coli (APC) is inhibited by signaling, and β-CATENIN gradually accumulates in the cytoplasm (Jessen et al, 2008) and migrates to the nucleus, where it interacts with the transcription factor T cell factor/lymphoid enhancer factor (TCF/LEF) combined to achieve the purpose of regulating cell fate, migration, and tissue configuration (Logan and Nusse, 2004; Xue et al, 2021). When the Wnt ligands are not activated, the destruction complex is able to continuously degrade β-CATENIN to maintain a lower cytoplasmic β-CATENIN protein level and inhibit signal transmission (Torres et al, 2019).
In the adhesive pool, β-CATENIN acts as the core of the adherens junctions (Oda and Takeichi, 2011) and regulates the aggregation of cadherin by directly binding to the cytoplasmic domain of cadherin and the actin-binding protein α-CATENIN (Nelson, 2008), maintaining cell–cell junctions, tissue structural integrity, and homeostasis in vivo (Garcia et al, 2018; van der Wal and van Amerongen, 2020).
Evolutionary Wnt/β-CATENIN Signaling and Cell Adhesion
From the beginning of life, protists were constantly diversifying. Organisms consisting of only single cells gradually evolved into complex multicellular organisms. The emergence of multicellular organisms is one of the major evolutionary transitions in the history of life (Sebé-Pedrós et al, 2017). During the evolution from protists to animals, the connection between the Wnt signaling and adherens junctions has deepened (Schneider et al, 2003). So far, adherens junctions have only been observed in metazoans, and the evolution of adherens junctions are still a hotspot of research today (Grimson et al, 2000). Not only actin-associated intercellular junctions were found in the protist Dictyostelium discoideum, but a gene encoding a homolog of β-CATENIN, AARDVARK was also characterized. AARDVARK can not only form a junction complex but also transmit cellular signals independently. Such results suggest that β-CATENIN has dual roles of cell signaling and cell–cell adhesion before the origin of metazoans (Grimson et al, 2000).
Apart from that, epithelia are an essential characteristic of metazoans. The cell–cell adhesion complex composed of cadherin, Wnt signal protein β-CATENIN, and α-CATENIN is the basis for maintaining epithelial structural integrity and functional polarization. The nonmetazoan Dictyostelium, although lacking the cadherin homolog, has a α-CATENIN homolog, which binds β-CATENIN-related proteins. Thus, catenins play a role in cell polarity long before Wnt signaling and canonical cadherins (Dickinson et al, 2011). The choanoflagellate Monosiga brevicollis is the closest known protist to metazoans and has cadherins that are otherwise expressed only in metazoans (Abedin and King, 2008; King, 2004; King et al, 2003). Sponges are the oldest extant metazoans, and Amphimedon queenslandica differs from M. brevicollis cadherins in that A. queenslandica has metazoan-specific domains of the canonical cadherin and seven-transmembrane cadherin subfamily (Srivastava et al, 2010).
β-CATENIN of the two-cell-layer metazoan Nematostella vectensis is stably expressed along the oral–aboral axis, translocating to the nucleus of the cells at gastrulation and specifying the endoderm (Wikramanayake et al, 2003). Wnt signaling is tightly linked to axis formation in the two-layer metazoan Hydra with a developed adhesion system (Hobmayer et al, 2000). Analyses of β-CATENIN in Drosophila, Caenorhabditis elegans, and vertebrates revealed that a single β-CATENIN was capable of both adhesion and signaling functions in metazoans (Schneider et al, 2003). Wnt signaling and cell adhesion are often active in the same process, and they form a crosstalk by regulating and sharing components with each other (Sylvie et al, 2011). However, the adhesion and Wnt signaling of C. elegans are performed by different β-CATENIN-related genes (Korswagen et al, 2000).
Separation of β-CATENIN Transcriptional Pool and Adhesive Pool
We already know the dual role of β-CATENIN for cadherin-based adherens junctions and Wnt signaling, but how this ensures the independence of the transcriptional and adhesive pools is not well understood (Valenta et al, 2012). The prevailing idea now is that the structural composition of β-CATENIN guarantees its dual function. The structure of β-CATENIN is mainly divided into three regions: N-terminal domain, C-terminal domain, and central core, wherein the central core region is composed of 12 armadillo repeat sequences (R1–R12) (Schneider et al, 2003).
Many molecules that need to bind to β-CATENIN to function have overlapping binding sites on β-CATENIN. To ensure the functional independence of the transcriptional pool and the adhesive pool, these molecules do not bind to β-CATENIN at the same time (Valenta et al, 2012; van Hengel et al, 2013). For example, the complexes formed by E-CADHERIN and β-CATENIN, LEF-1 and β-CATENIN are mutually exclusive (Orsulic et al, 1999). Those with structural regions responsible for cell adhesive function but with N-terminal and C-terminal mutations β-CATENIN transgenic mice do not produce abnormal cell adhesion defects (Valenta et al, 2011).
It has also been reported that by generating domain-deleted mutants of Drosophila, researchers found that β-CATENIN homologous genes play a role in signal transduction early in neural development and that β-CATENIN homologous genes affect adherens junctions later in development (Loureiro and Peifer, 1998). Therefore, the structure of β-CATENIN plays a key role in the separation of the two pools.
Another opinion now exists, because different molecular forms of β-CATENIN have different functional preferences, which determine whether it plays a role in adhesion junctions or Wnt signaling. When the Wnt signaling is activated, the COOH end of β-CATENIN selectively binds to TCF but not to cadherin through an intramolecular folding mechanism. Phosphorylation of cadherin can reverse the binding of β-CATENIN to TCF. However, β-CATENIN binds mainly to cadherin as a β-CATENIN–α-CATENIN dimer, a different molecular form of β-CATENIN (Gottardi and Gumbiner, 2004). In addition to this, unmodified β-CATENIN, its N-terminal and C-terminal is able to bind and function with the armadillo repeat domain. However, when β-CATENIN undergoes posttranslational modification, the N-or C-terminus of β-CATENIN is placed in a specific conformation that regulates the binding of specific molecules to β-CATENIN (Xing et al, 2008).
In C. elegans, β-CATENIN homologous genes play different roles. The roundworm C. elegans has at least four β-CATENIN homologous genes, including BAR-1, HMP-2, SYS-1, and WRM-1. HMP-2 is dedicated to adherens junctions. BAR-1, WRM-1, and SYS-1 have the function of transcriptional activation of β-CATENIN (Korswagen et al, 2000; Liu et al, 2008).
Crosstalk Between β-CATENIN-Mediated Cell Adhesion and the Wnt Signaling
Although there is evidence that the adherens junctions and Wnt signaling are independent of each other, there is still a lot of evidence that the adherens junctions and the Wnt signaling crosstalk. Overexpression of all three cadherins in Xenopus embryos resulted in the damage to the dorsal axis of the developing embryo, which was rescued by injection of β-catenin mRNA (Heasman et al, 1994). In addition to this, overexpression of C-CADHERIN depletes β-CATENIN from the cell membrane and cytoplasm and inhibits the ability of β-CATENIN to transmit signals (Fagotto et al, 1996). Overexpression of E-CADHERIN antagonizes β-CATENIN/LEF-1-mediated transactivation (Orsulic et al, 1999). In contrast, overexpression of γ-CATENIN-induced transactivation of Lef-1 (Simcha et al, 1998). ADAM10 can indirectly affect the Wnt signaling by affecting the interaction between E-CADHERIN and β-CATENIN (Guerra et al, 2021; Li et al, 2017).
If the two disparate functions of maintaining adherens junctions and mediating the Wnt signaling are performed by the same protein, and these two functional pools of β-CATENIN are separated from each other, then how to explain the crosstalk between adherens junctions and the Wnt signaling becomes the focus (van der Wal and van Amerongen, 2020).
Crosstalk caused by competitive binding with β-CATENIN
To maintain the integrity of adhesion connection and to transmit the Wnt signal, β-CATENIN has different interacting partners, and these molecules compete with each other to maintain the balance and crosstalk between the two pools (Table 1). By overexpressing of N-CADHERIN and IL-2 receptor-cadherin chimera in SW480 cells, cadherin derivatives prevent β-CATENIN degradation by forming a complex with β-CATENIN. The localization of β-CATENIN is transferred from the nucleus to the plasma membrane, reducing the transactivation of LEF-1 reporter gene. N- or E-CADHERIN in the nucleus can also block the interaction between β-CATENIN and LEF-1 and inhibit the transactivation mediated by LEF-1. Therefore, cadherin can inhibit the degradation of β-CATENIN and its transactivation ability in both cell membrane and nucleus (Sadot et al, 1998). E-CADHERIN and LEF-1 compete for the binding to β-CATENIN. The deletion of E-CADHERIN in embryonic stem (ES) cells promotes the binding of β-CATENIN to LEF-1 and activates the Wnt pathway (Orsulic et al, 1999).
An Overview of Regions and Residues That Determine β-CATENIN Binding Partner
APC, adenomatous polyposis coli; TCF/LEF, T cell factor/lymphoid enhancer factor.
It has also been proposed that E-CADHERIN, as a cell adhesion protein, has an adhesion-independent effect on tumors. It can deplete the β-CATENIN transcriptional pool (Gottardi et al, 2001). In FosER cells, β-CATENIN, p120-CATENIN, and E-CADHERIN can form a tight complex located on the cell membrane. When the cells lose epithelial polarity, the expression of E-CADHERIN is completely lost, the level of β-CATENIN is significantly decreased, and the expression pattern of p120-CATENIN isoform changes and activates the transcriptional activity of LEF-1. These changes can be saved by overexpression of exogenous E-CADHERIN. Therefore, epithelial mesenchymal transformation (EMT) in FosER cells is closely related to β-CATENIN signaling (Eger et al, 2000). Overexpression of membrane-localized β-CATENIN increases the accumulation of free β-CATENIN in the nucleus after interaction with APC.
In response, cadherin binds to free β-CATENIN and inhibits Wnt signaling (Miller and Moon, 1997). E-CADHERIN also competes with APC for binding to β-CATENIN, thereby mediating the interaction of E-CADHERIN and the APC complex with the cytoskeleton (Hülsken et al, 1994). Overexpression of E-CADHERIN can rescue intermittent cyclic mechanical tension (ICMT)-induced decreased interaction between E-CADHERIN and β-CATENIN to increase cartilage gene expression (Xu et al, 2016). It can also avoid binding to β-CATENIN by binding to CCT5, thus promoting nuclear transfer of β-CATENIN and enhancing the Wnt signaling (Li et al, 2022). The effects of E-CADHERIN treatments are similar to those of GSK3 inhibitors (Fan et al, 2018). Thus, the competitive binding of chaperones to β-CATENIN will balance the functional stability of the two pools.
Phosphorylation-mediated crosstalk
To maintain the balance between cell adhesion and the Wnt signaling, β-CATENIN forms a cadherin–catenin complex with cadherin. The cadherin–catenin complex, the disruption complex in the Wnt signaling, and the TCF transcription system act in concert and are continuously connected, with phosphorylation playing a large role. It not only ensures the integrity of the structure and function of the cadherin–catenin complex, but also affects the signaling of the Wnt signaling (Heuberger and Birchmeier, 2010; Nelson and Nusse, 2004). The phosphorylation of both adherens junction-related protein and β-CATENIN can affect the balance of the two pools. For example, the phosphorylation of p120-CATENIN and E-CADHERIN is closely related to the functional integrity of β-CATENIN. The kinase Src-mediated phosphorylation of β-CATENIN at tyrosine 654 reduces the binding capacity of cadherin. Phosphorylation at β-CATENIN tyrosine 142 mediated by kinase Fer and Met reduces α-CATENIN binding. Phosphorylation of β-CATENIN at position 489 affects the binding ability of N-CADHERIN (Valenta et al, 2012).
Crosstalk mediated by β-CATENIN phosphorylation
The weakened interaction of β-CATENIN with cadherin and α-CATENIN caused by the phosphorylation of β-CATENIN is the key point for β-CATENIN to switch from an adherens junction mode to a signaling mode (Fig. 1). Control of cadherin by phosphorylation at Tyr654 of β-CATENIN promotes the transition of β-CATENIN to a signaling mode (Brembeck et al, 2006; Valenta et al, 2012).

Crosstalk mediated by β-CATENIN phosphorylation. Affects the cadherin–β-CATENIN complex by phosphorylation of β-CATENIN at Tyr654, promoting the conversion of β-CATENIN to a signaling mode. The kinases Src, Met, and Trk can all cause the phosphorylation of β-CATENIN at position 654. TG2, PrP-1, HMGB1, AGEs, Ouabain, UPA promote the phosphorylation of kinase Src; Prx2, SHP-1 inhibit the phosphorylation of kinase Src; SHP-1 can also inhibit the signaling of β-CATENIN by increasing the activity of GSK3; NTs through Trk causes phosphorylation of β-CATENIN at position 654; HGF causes phosphorylation of β-CATENIN at position 654 by Met. Phosphorylation of β-CATENIN at position 142 disrupts the α-CATENIN–β-CATENIN complex and promotes signaling. Kinases Fer, Met and Fak can all cause phosphorylation of β-CATENIN at position 654, HGF and UVA activate kinase Met; HMGB1 and AGEs activate kinase Fak; CONNEXIN43 mediates the dissociation of α-CATENIN–β-CATENIN complex through Fer kinase. β-CATENIN Tyr489 can also affect adherens junctions by affecting N-CADHERIN. ROBO binds to Slit to recruit cables to ABL and bind to β-CATENIN, followed by phosphorylation of β-CATENIN Tyr489 to disrupt the binding between β-CATENIN and N-CADHERIN. ABL, c-Abl tyrosine kinase; AGE, advanced glycation end product; GSK3, glycogen synthase kinase 3; HGF, hepatocyte growth factor; HMGB1, high mobility group box-1 protein; ROBO, Roundabout; Prx2, peroxiredoxin-2.
Kinase Src is critical to this process. The multifunctional enzyme tissue transglutaminase (TG2) may serve as an example. In ovarian cancer cells, Tg2 tends to recruit and bind to c-Src to phosphorylate β-CATENIN at Tyr654, thereby dissociating β-CATENIN from the cadherin–catenin complex and enhancing Wnt/β-CATENIN signaling, together with TG2 to stimulate OC cell proliferation (Condello et al, 2013). At the upstream of Src family kinases, Fyn and Yes, PrP inhibits the endocytosis and degradation of the E-CADHERIN–β-CATENIN complex by affecting Src (Sempou et al, 2016).
Advanced glycation end products (AGEs) and high mobility group box-1 proteins (HMGB1) can disrupt the permeability of vascular endothelial cells by causing the dissociation of E-CADHERIN–β-CATENIN through the Src kinase (Weng et al, 2021; Weng et al, 2019). In contrast, tyrosine phosphatase SHP-1 can negatively regulate the nuclear transcriptional activity of β-CATENIN. SHP-1 significantly reduced the transactivation potential of β-CATENIN by inhibiting Src-induced β-CATENIN tyrosine phosphorylation at positions 86 and 654. In addition, Pervanadate inhibits the negative effects of tyrosine phosphatase (Simoneau et al, 2011).
The transforming growth factor-β1 (TGF-β1) pathway is tightly associated with phosphorylation at Tyr654 of β-CATENIN, and the phosphorylation caused by TGF-β1 leads to the formation of the p-β-CATENIN-Y654/p-Smad2 transcription complex, which in turn upregulates the expression of α3 integrin-dependent integrin-linked kinase and decreases the expression of E-CADHERIN, leading to tissue damage, loss of kidney tubular epithelial cell integrity, and fibrosis of lung epithelial cells. Autophagy is important for activation of the TGF-β1 pathway (Kim et al, 2009; Pang et al, 2016; Zheng et al, 2016).
The force of 6pN physiological mechanical strain can also release β-CATENIN from the Y654-β-CATENIN/D665-E-CADHERIN complex, initiating the activation of the Wnt signaling (Röper et al, 2018). The Src and ERK signaling pathways are negatively regulated by the antioxidant enzyme peroxiredoxin-2 and positively regulated by the plant-derived toxin Ouabain to coordinate adherens junctions and signaling (Castillo et al, 2019; Lee et al, 2013). In addition to kinase Src, the receptor tyrosine kinases, Met and Trk, also have a significant effect on the phosphorylation of β-CATENIN Tyr654. Epidermal growth factor receptor (EGFR) activation by UVA causes disruption of the E-CADHERIN/α-CATENIN/β-CATENIN complex and EGFR-dependent transfer of β-CATENIN localization (Jean et al, 2009). Neurotrophins can affect axonal growth and differentiation through Trk receptors (David et al, 2008).
Similar to E-CADHERIN, N-CADHERIN is also affected by β-CATENIN phosphorylation at Tyr654, which in turn leads to the regulation of presynaptic vesicle dynamics in synaptic connections and the maintenance of synaptic communication in neurons (Chen et al, 2017). Urokinase-type plasminogen activator-triggered phosphorylation also induces wound healing in astrocytes (Diaz et al, 2021).
Unlike β-CATENIN Tyr654 phosphorylation, β-CATENIN Tyr142 affects adherens junctions by disrupting the binding between β-CATENIN and α-CATENIN. For example, hepatocyte growth factor (HGF) can shed β-CATENIN from the cell membrane to the nucleus and bind to BCL9 by activating kinase Met to promote transcription and regulate embryonic mesoderm development and axogenesis (Brembeck et al, 2004; David et al, 2008; Howard et al, 2011). The kinase Fak also plays a key role in phosphorylation of β-CATENIN Tyr142.
AGEs not only disrupt the binding between β-CATENIN and α-CATENIN through Fak, but also activate the Wnt pathway to increase the transcription of the target gene ADAM10, resulting in the shedding of VE-CADHERIN and further damage to cell adhesion (Weng et al, 2021). Cytoskeleton damage caused by HMGB1 is similar to that of AGEs (Weng et al, 2019). Although phosphorylation of β-CATENIN Tyr142 is generally believed to affect adherens junctions, CONNEXIN43 can increase endothelial cell monocytic adhesion through kinase Fer-mediated phosphorylation of Tyr142 (Chang et al, 2014).
β-CATENIN Tyr489 can also affect adherens junctions by affecting N-CADHERIN. Roundabout (ROBO) binds to Slit to recruit cables to c-Abl tyrosine kinase (ABL) and bind to β-CATENIN, followed by phosphorylation of β-CATENIN Tyr489 to disrupt the binding between β-CATENIN and N-CADHERIN, release N-CADHERIN, and affect adhesion (Rhee et al, 2007).
Crosstalk mediated by adherens junction-related protein phosphorylation
In addition to β-CATENIN, phosphorylation of adherens junction-related proteins are equally important for the crosstalk of the two pools (Fig. 2). As part of the Wnt pathway, phosphorylation of CK1ɛ regulates the normal transmission of Wnt signaling in two ways. It not only binds and phosphorylates the protein p120-CATENIN used to regulate the stability of E-CADHERIN, and promotes p120-CATENIN to accept the stimulation of Wnt3a, but also phosphorylates β-CATENIN to reduce the affinity with E-CADHERIN, thereby regulating the nuclear β-CATENIN metastases, and ultimately causing aberrant activation of the Wnt pathway (Casagolda et al, 2010). Another CK1 kinase isoform, CK1α, plays a role subsequent to CK1ɛ. After activation of Wnt3a, CK1α forms a complex with Lrp5/6 and p120-CATENIN, which is involved in the release of p120-CATENIN and E-CADHERIN (Del Valle-Pérez et al, 2011), and the released P120-CATENIN interacts with VAV2 to activate RAC1, which is required for the Wnt signaling (Valls et al, 2012).
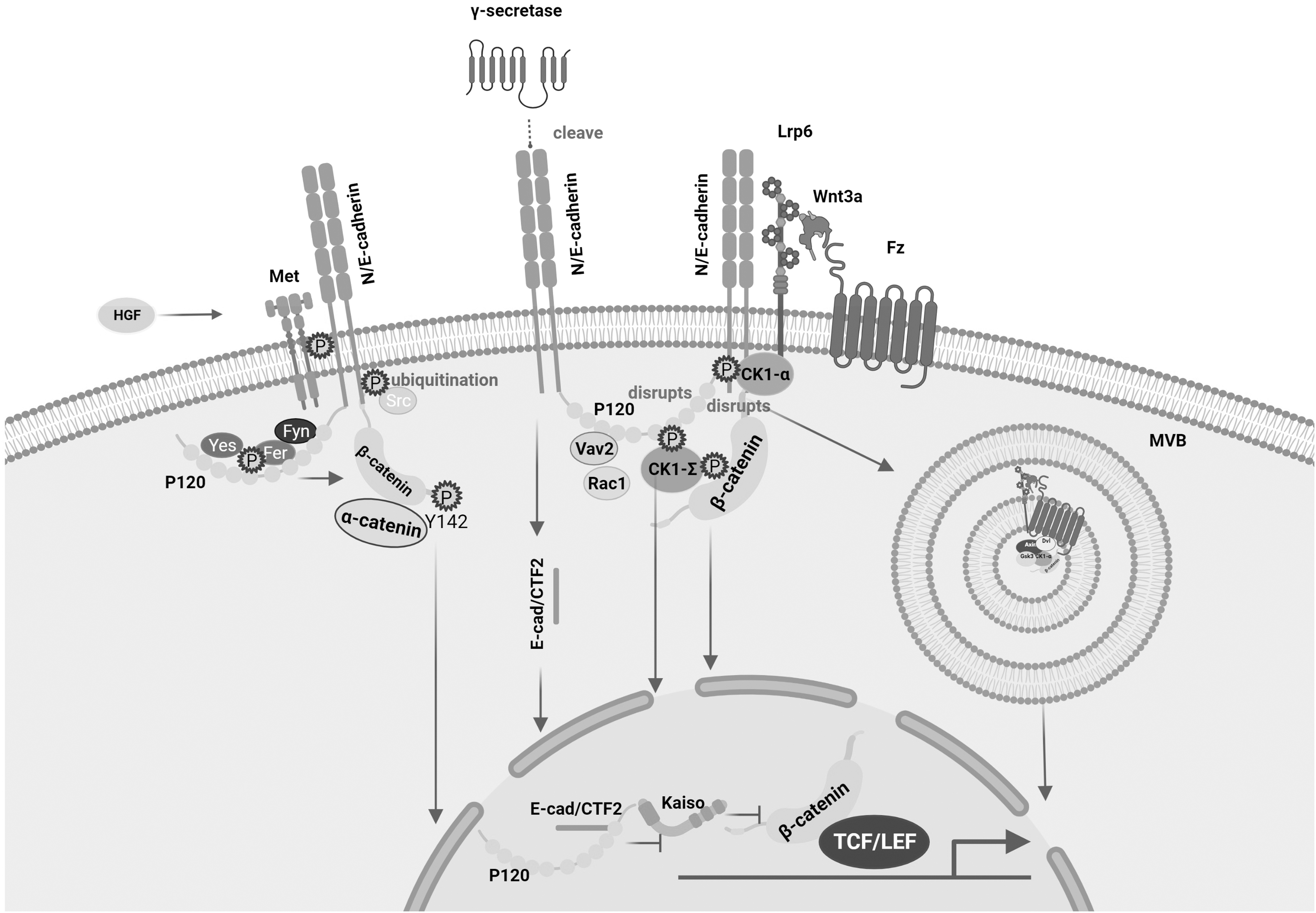
Crosstalk mediated by adherens junction-related protein phosphorylation. CK1ɛ not only binds and phosphorylates p120-CATENIN, a protein used to regulate E-CADHERIN stabilization, but also phosphorylates E-CADHERIN to reduce its affinity for β-CATENIN, thereby regulating β-CATENIN nuclear transfer; Another isoform of CK1 kinase, CK1α, forms a complex with Lrp5/6 and P120-CATENIN, which is involved in the release of P120-CATENIN and E-CADHERIN, and the released P120-CADHERIN interacts with VAV2 to activate RAC1. P120-CATENIN phosphorylation destroys the N/E-CADHERIN–P120-CATENIN complex, which facilitates the internalization of Wnt signaling into MVB to promote transcription; the nucleus of E-CAD/CTF2 obtained by γ-SECRETASE cleavage Metastasis affects the Wnt pathway by affecting the p120-KAISO pathway; p120 phosphorylation enhances the binding ability of Fer/Fyn-p120-CATENIN and E-CADHERIN. Phosphorylation of E-CADHERIN by Src kinase induces DNF43-mediated degradation of E-CADHERIN by ubiquitination; HGF can induce phosphorylation of E-CADHERIN through the kinase Met, resulting in nuclear transfer of β-CATENIN. Lrp5/6, lipoprotein receptor-related proteins 5 and 6; MVB, multivesicular bodies.
P120-CATENIN phosphorylation disrupts the N/E-CADHERIN/p120-CATENIN complex, favoring the internalization of Wnt signaling into multivesicular bodies to promote transcription (Vinyoles et al, 2014). In addition, P120 promotes the nuclear transfer of E-CAD/CTF2 generated by cleavage of E-CADHERIN by γ-SECRETASE, thereby affecting the p120-KAISO-mediated pathway and thus the Wnt signaling (Ferber et al, 2008). We have already mentioned that Fer and Fyn as tyrosine kinases can phosphorylate β-CATENIN Tyr142 and disrupt the binding between β-CATENIN and α-CATENIN, the activation of Fer and Fyn promoted by the phosphorylation of p120 induced by the oncogene K-RAS enhancing the binding of Fer/Fyn-p120-CATENIN to E-CADHERIN, and these results emphasize the importance of p120-CATENIN in adherens junctions (Piedra et al, 2003).
Phosphorylation of E-CADHERIN can also promote the activation of the Wnt signaling, and phosphorylation of E-CADHERIN by Src promotes DNF43-mediated ubiquitin degradation of E-CADHERIN, resulting in β-CATENIN nuclear transfer (Zhang et al, 2019). HGF can induce E-CADHERIN phosphorylation through the kinase Met (Matteucci et al, 2006).
Thus, phosphorylation is critical for balancing the Wnt signaling and cell adhesion.
Wnt/β-CATENIN signaling target genes and EMT
Phosphorylation regulates the homeostasis of the Wnt signaling and adherens junctions, and in the process of this homeostasis, there is also a universal and important physiological phenomenon, EMT. EMT is a reversible process that is integral to development and occurs in a variety of cells and tissues, as evidenced by the loss of polarity in epithelial cells and the acquisition of mesenchymal cellular properties, with a decrease in E-cadherin expression in cells and an increase in N-cadherin expression (Cho et al, 2019). Many pathways are involved in the process of EMT, of which the Wnt signaling is a very important part (Huang et al, 2022). The influence of the Wnt signaling on EMT is mainly reflected in two aspects: first, the transcription factors SNAIL, SLUG, and TWIST targeted by Wnt/β-CATENIN regulate the transcription and protein levels of E/N-CADHERIN; second, the weakening and enhancement of adherens junctions by Wnt/β-CATENIN-targeted genes (Fig. 3) (Heuberger and Birchmeier, 2010).
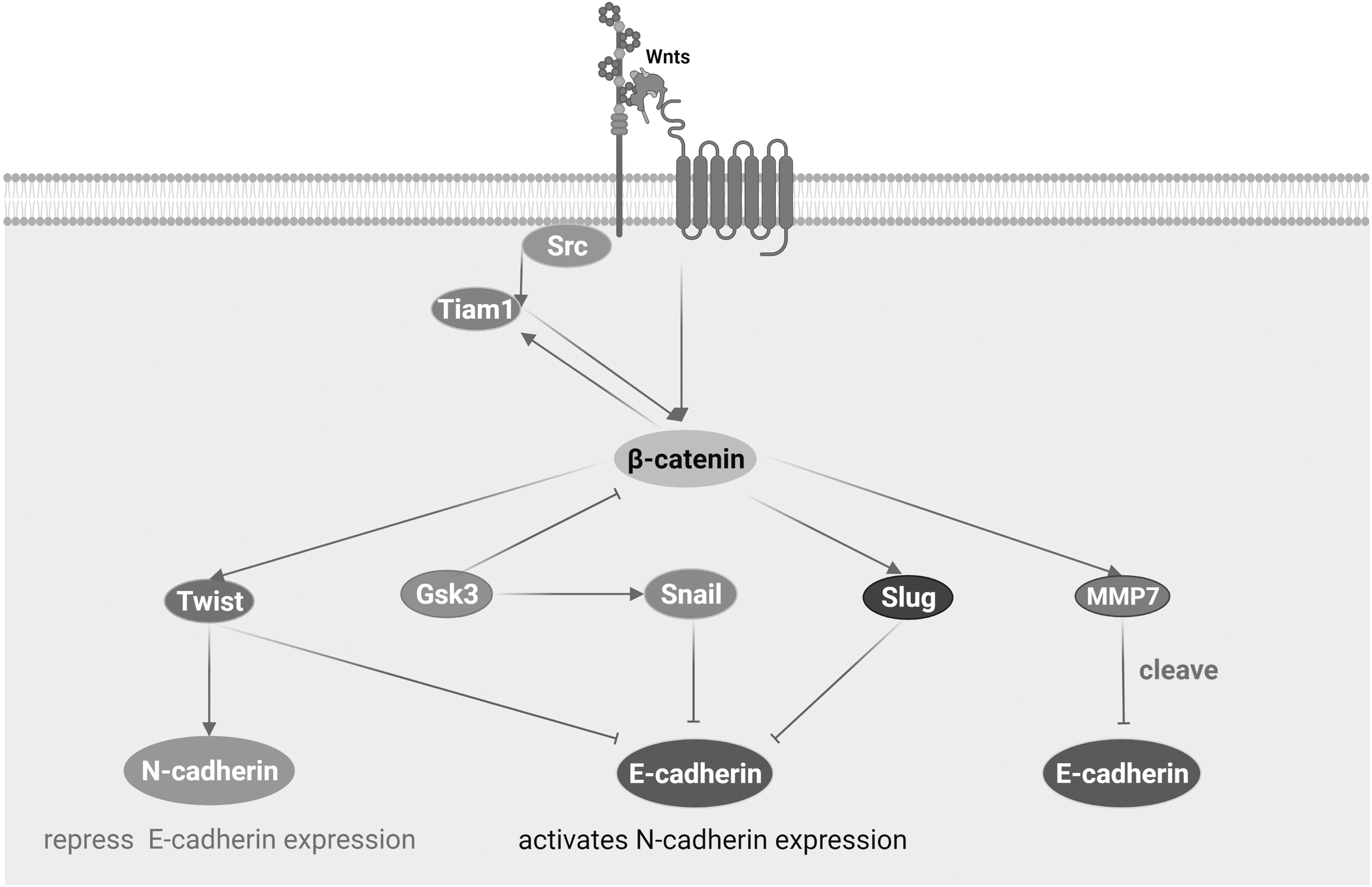
Wnt/β-CATENIN signaling target genes and EMT. During EMT, transcription factors SNAIL and SLUG can target and inhibit the transcription of E-CADHERIN, while TWIST can not only inhibit the transcription of E-CADHERIN, but also enhance the transcription of N-CADHERIN. SNAIL needs to be stabilized by GSK3, which is a substrate of GSK3 and is affected by ubiquitination and phosphorylation of GSK3. MMP7 acts as a target gene and affects EMT by cleaving E-CADHERIN. TIAM1 is both a Wnt target gene and reversely affects the Wnt signaling, and phosphorylation of TIAM1 by Src is required for Src to disrupt adherens junctions. EMT, epithelial–mesenchymal transformation; MMP7, matrilysin.
SNAIL and SLUG are indispensable in the process of EMT. They can target and affect the transcription of E-CADHERIN and often play a role in cancer. For example, EFEMP2 is an extracellular matrix protein that inhibits the transcription of E-CADHERIN through Wnt/β-CATENIN, acting on SNAIL and SLUG, and inhibiting bladder cancer EMT (Zhou et al, 2019). ERBB3-binding protein 1 and sex comb on mid leg like-2, promoting the metastasis of melanoma cells as well as hepatoma cells according to similar principles (Bao et al, 2022; Du et al, 2021; Untiveros et al, 2021). Decreased SNAIL also causes increased E-CADHERIN expression to control cancer progression (Sun et al, 2021). It shows that the activation of SNAIL plays a positive feedback role on the Wnt signaling (Stemmer et al, 2008). Of course, the effect of these transcription factors on EMT is not only reflected in cancer, but also in the intervention of SNAIL in rheumatoid arthritis (Shen et al, 2021) and diseases such as endometriosis (Xiong et al, 2019).
It is worth noting that SLUG has a binding site for LEF, so it can directly accept the signal of β-CATENIN/TCF to regulate the transcription of E-CADHERIN.
However, SNAIL needs to be stabilized by GSK3. SNAIL is the substrate of GSK3 and plays a role by ubiquitination and phosphorylation of GSK3 (Zhou et al, 2004). SNAIL and SLUG can target and inhibit the transcription of E-CADHERIN, whereas TWIST can not only inhibit the transcription of E-CADHERIN, but also enhances the transcription of N-CADHERIN. As a bHLH transcription factor, it was originally discovered in Drosophila and identified as an important molecule for inducing mesoderm formation (Simpson, 1983), and was later confirmed to be involved in EMT through the opposite regulation of N-CADHERIN and E-CADHERIN (Oda et al, 1998). TWIST is necessary for inducing cancer cell metastasis after E-cadherin deletion (Kong et al, 2021; Onder et al, 2008). Overexpressed TWIST can increase the expression of N-CADHERIN and decrease the expression of E-CADHERIN in gastric cancer cells, MKN45. Unexpectedly, the downregulation of TWIST inhibits the expression of N-CADHERIN, but failed to have any effect on the expression of E-CADHERIN (Yang et al, 2007).
EMT is also affected by the stability of E-CADHERIN. Unstable E-CADHERIN promotes the realization of EMT. Metalloproteinase matrilysin (MMP7), as the target gene of Wnt/β-CATENIN signaling, is involved in the cleavage of E-CADHERIN (Crawford et al, 1999). As a target gene, MMP7 upregulates cleavage of E-CADHERIN when the Wnt/β-CATENIN signaling is activated, so that it detaches from the cell membrane and promotes the spread of colon cancer, bladder cancer, breast cancer, and other cancer cells (Tao et al, 2019; Wang et al, 2020a; Wang et al, 2020b; Zhang et al, 2020). It is important to note that only PEA3 subfamily members cooperating with β-CATENIN can cause an upregulation of MMP7 transcription (Crawford et al, 2001). In addition to MMP7, the Wnt target gene TIAM1 is also involved in EMT.
TIAM1 is affected by RAC1 for Wnt/β-CATENIN signaling-mediated EMT, while TIAM1 is also a RAC1 activator. Phosphorylation of TIAM1 by kinase Src is required for Src to disrupt adhesive junctions (Liu et al, 2018; Malliri et al, 2006; Woodcock et al, 2009), and its ectopic expression enhances cell adhesion by increasing E-CADHERIN (Hordijk et al, 1997), thus TIAM1 can control tumor progression (Chapelle et al, 2020).
In conclusion, EMT is closely related to a crosstalk between cell adhesion and Wnt signaling.
γ-CATENIN and Wnt/β-catenin signaling pathway
For the exploration between cell adhesion and Wnt signaling, we need to pay attention to a very specific protein, γ-CATENIN, in addition to the aspects summarized above. γ-CATENIN is a homologous protein with a high degree of structural and functional similarity to β-CATENIN. Structurally, γ-CATENIN and β-CATENIN have similar structures and armadillo repeat sequences, which are common to CATENINs (Martin et al, 2009). Functionally, γ-CATENIN is also a multifunctional protein, which is able to act as a cell adhesion molecule not only for desmosomes and adherens junctions, but also to activate the Wnt signaling independently of β-CATENIN (Maeda et al, 2004). For adherens junctions, γ-CATENIN not only connects CADHERINs to α-CATENIN and F-ACTIN, but also binds CADHERINs competitively with β-CATENIN (Mahendram et al, 2013). When γ-CATENIN acts as a signaling molecule, it can bind to TCF4/LEF1 to activate the Wnt signaling.
Although it mediates weak transcriptional activity, the fact that γ-CATENIN is an important Wnt pathway signaling molecule still cannot be ignored (Maeda et al, 2004), and the main proteins in the Wnt signaling, AXIN, WNT1, and APC, can regulate its stability (Kodama et al, 1999; Papkoff et al, 1996). We now generally believe that when γ-CATENIN is overexpressed it can cause accumulation and nuclear translocation of β-CATENIN and induce transactivation of LEF1 (Simcha et al, 1998; Zhurinsky et al, 2000), which may be due to γ-CATENIN competing with β-CATENIN to bind β-TrCP in the ubiquitin–proteasome system, stabilizing β-CATENIN and driving β-CATENIN into the nucleus (Sadot et al, 2000; Salomon et al, 1997). However, there are still different views on the effects of reduced γ-CATENIN on organisms.
In terms of adherens junctions, it has been proposed that knockdown of γ-CATENIN does not cause abnormal adherens junctions in hepatocellular carcinoma (HCC) cells because the expression of β-CATENIN is compensatively increased and the binding of β-CATENIN–E-CADHERIN will not change (Wickline et al, 2013). In γ-CATENIN cardiorestricted knockout mice, β-CATENIN also upregulates expression and compensates for adherens junctions (Li et al, 2011).
However, it has been mentioned that the reduction of γ-CATENIN in mouse embryonic stem cells (mESCs) did not cause a compensatory increase in the expression of β-CATENIN (Mahendram et al, 2013).
In terms of Wnt signaling, knockdown of γ-CATENIN caused activation of Wnt signaling in HCC cells and γ-CATENIN-deficient zebrafish (Martin et al, 2009; Wickline et al, 2013), but not in mESCs (Mahendram et al, 2013). In γ-CATENIN cardiorestricted knockout mice, some authors suggested that the rise in β-CATENIN expression only compensated for adherens junctions without effect on the Wnt signaling (Li et al, 2011a), while some authors suggested that γ-CATENIN knockdown activated β-CATENIN/TCF (Li et al, 2011b).
When β-CATENIN expression was reduced, γ-CATENIN was able to upregulate expression and replace β-CATENIN in HCC cells, mESCs, pancreatic adenocarcinoma cells, and renal tubules conditional knockout mice to maintain adherens junctions by binding to E-CADHERIN, a phenomenon that was independent of the Wnt signaling (Mahendram et al, 2013; Olsen et al, 2014; Wickline et al, 2013; Zhou et al, 2012). Meanwhile, the increase of γ-CATENIN expression in HCC cells could only occur when the expression of β-CATENIN protein was decreased, and blocking the transactivation of Wnt cannot increase the expression of γ-CATENIN (Wickline et al, 2013). However, it has also been suggested that in transgenic mouse models of ErbB2-induced mammary tumorigenesis, the defect in β-CATENIN can be replaced by γ-CATENIN in both adherens junctions and transcription (Bui et al, 2017). Thus, γ-catenin is also closely related to the two-pool crosstalk of β-catenin.
Crosstalk between cell adhesion and the Wnt/β-CATENIN signaling in disease
EMT is closely related to cancer, and cancer naturally becomes a ubiquitous phenomenon in the interaction of cell adhesion and the Wnt signaling. However, in addition to cancer, there are many diseases that can reflect the crosstalk of cell adhesion and the Wnt signaling, the most representative of which are Alzheimer's disease and glaucoma (Uemura et al, 2006; Wecker et al, 2013).
Synapses allow neurons to perform physiological functions, and N-CADHERIN is a widely present adhesion molecule at synapses and necessary for synaptic excitation in the hippocampus. Presenilin1 (PS1), as an important protein involved in cell development, is also localized at synapses, and its loss of function at synaptic sites is closely related to Alzheimer's disease. PS1 can form a complex with β-CATENIN and N-CADHERIN. N-CADHERIN is the key to the formation of the PS1/β-CATENIN complex, which is also a prerequisite for Alzheimer's disease (Uemura et al, 2006). Under the stimulation of glutamate, PS1 and γ-SECRETASE altogether cleave N-CADHERIN to generate fragmented N-CAD/CTF2 that is translocated to the nucleus. N-CAD/CTF2 can not only directly mediate the movement of β-CATENIN to the nucleus but also inhibit the phosphorylation of β-CATENINSer33/37/Thr41 in promoting the accumulation of β-CATENIN. Of course, the dissociation of the N-CADHERIN–β-CATENIN complex also caused cytoplasmic accumulation of β-CATENIN.
Therefore, PS1 activates the Wnt/β-CATENIN signaling from these three aspects (Uemura et al, 2006). PS1 can also cleave E-CADHERIN with γ-SECRETASE, dissociate the E-CADHERIN–catenin complex, and dissociate E-CADHERIN, β-CATENIN, and α-CATENIN from the cytoskeleton. Intracytoplasmic β-CATENIN levels promote the Wnt/β-CATENIN signaling (Marambaud et al, 2002; Serban et al, 2005). Glaucoma is a disease characterized by neurodegeneration. To avoid glaucoma, K-CADHERIN maintains the trabecular meshwork and IOP through the Wnt/β-CATENIN signaling (Webber et al, 2018). In addition to the Wnt/β-CATENIN signaling, the cytokine TGF-β2 is closely related to enhanced cadherin-mediated intercellular adhesion and β-CATENIN level in trabecular meshwork cells. Therefore, for the treatment of glaucoma, not only the Wnt/β-CATENIN signaling, but also TGF-β2 deserves our particular attention (Wecker et al, 2013).
Conclusions and Perspectives
As a classical molecule that is crucial to development, the exploration of β-CATENIN has ever increased. Since independent studies of the Wnt/β-CATENIN signaling and cell adhesion, β-CATENIN-induced crosstalk has continued to be discovered, and it is clear that there is a link between them. How β-CATENIN coordinates the balance between its complex functions and how it affects the stability of organisms has become a hot research topic today. This article links the Wnt/β-CATENIN signaling with cell adhesion from five aspects: the competitive binding of β-CATENIN chaperone molecules, the phosphorylation of β-CATENIN, EMT, the β-CATENIN homologous protein γ-CATENIN and disease, so that they form a tight network model and explain the basic mechanism of disease emergence. But for the current work, in addition to the central role of β-CATENIN, we believe more pathways and the coordination of signaling molecules should be involved. β-CATENIN cannot be an isolated individual, and many pathways related to growth and development should be considered, such as TGF-β2 in glaucoma disease.
In addition, we need to understand the preferred molecules involved in β-CATENIN signaling, such as the kinase Src, the kinase Met, BCL2, etc. Only by really considering these factors can we gain a deeper understanding of our confusion. The relationship between aberrant activation of the Wnt/β-CATENIN signaling and disruption of cell adhesion is the key point for analyzing how some diseases occur. The exploration of the above crosstalk will provide a new platform for disease investigation. The understanding of the crosstalk will also provide new clinical application for the diagnoses and treatment of diseases in the future. It may also provide assistance in the development of more meaningful therapeutic medical approaches in the future.
Footnotes
Acknowledgments
The authors greatly appreciate all members of the Sperm Laboratory in Zhejiang University for their helpful suggestions and discussions on this study. Dr. Hans-Uwe Dahms is acknowledged for his critical reading of the final draft of the article.
Authors' Contributions
D.-X. Liu participated in the selection and collection of references, original draft, and figure preparation. W.-X.Y. and S.-L.H. participated in the conception, the structured outline preparation, the article revising, and the final revision of the article.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported in part by the National Natural Science Foundation of China (No. 32072954, 32102786, and 32270555).