
Abstract
The role of ferroptosis in human acute lymphoblastic leukemia and its possible molecular mechanisms of action are still unknown. In this study, harvested Molt-4 cells were exposed to different concentrations of erastin, and their proliferation capacity was tested by using the cell counting kit-8 assay. Lipid peroxidation levels were detected through flow cytometry. Mitochondrial alterations were observed through transmission electron microscopy. The expression levels of SLC7A11, glutathione peroxidase 4 (GPX4), and mitogen-activated protein kinase (MAPK) were detected by using quantitative real-time PCR and Western blot analysis. This study found that erastin inhibited the growth of Molt-4 cells. This inhibitory effect could be partially reversed by the ferroptosis inhibitor Ferrostatin-1 and the p38 MAPK inhibitor. The mitochondria of Molt-4 cells treated with erastin shortened and condensed. Compared with those in the control group, the levels of reactive oxygen species and malondialdehyde had increased, whereas the levels of glutathione had decreased in the treatment group. The treatment of Molt-4 cells with erastin decreased the levels of SLC7A11 and GPX4 mRNA and increased the expression levels of p38 MAPK, extracellular signal–regulated kinase (ERK), and c-Jun N-terminal kinase. These findings suggested that erastin caused the ferroptosis of Molt-4 cells. This process may be correlated with the inhibition of the cystine/glutamate antiporter system and GPX4 and the activation of p38 MAPK and ERK1/2.
Introduction
Acute lymphoblastic leukemia (ALL) is one of the most common malignant tumors of the blood system. Although the survival rate of patients with childhood ALL is close to 90% (Hunger et al., 2012; Inaba et al., 2013), the relative survival rates of patients aged 15–69 years in Germany and the United States are only 43% and 35%, respectively (Pulte et al., 2014). These epidemiological data emphasize that the thorough exploration of new therapeutic targets is the primary task in the research on ALL treatment.
Reports have shown that many forms of cancer cells develop into iron overload (Bystrom and Rivella, 2015). Epidemiological studies have found that patients with ALL suffer from high serum iron and excess intracellular iron before and after receiving chemotherapy (Olcay et al., 2014). Iron overload can stimulate the growth and proliferation of cancer cells and thus has an important value in the progression of leukemia (Richardson et al., 2009; Kovacevic et al., 2011). Moreover, iron overload plays a vital role in ferroptosis. Wang et al. (2022) found that the regulation of ferroptosis in leukemic cells could improve the effect of chemotherapy in patients with acute myeloid leukemia (AML). In addition to this, ferroptosis suppressor protein 1 (FSP1) is a ferroptosis protective mechanism. However, in ALL cell lines, FSP1 is silenced by DNA methylation, making it susceptible to ferroptosis (Pontel et al., 2022). These findings suggested that triggering ferroptosis might be an effective approach to the treatment of ALL.
In 2003, researchers discovered that in various cancer cell lines, erastin could trigger ferroptosis, a nonapoptotic form of cell death (Dolma et al., 2003; Yagoda et al., 2007; Yang and Stockwell, 2008; Dixon et al., 2012). In contrast to autophagy and apoptosis, ferroptosis is characterized by the accumulation of iron-dependent lethal lipid peroxides (Li and Li, 2020). Erastin could accumulate reactive oxygen species (ROS) of leukemia cells by inhibiting the cystine/glutamate antiporter system (System
It has been shown that mitogen-activated protein kinase (MAPK) is a downstream signaling of ROS, and that ROS accumulation leads to activation and phosphorylation of MAPK pathway members, which in turn induces apoptosis (Mantzaris et al., 2016). Subsequently, Hattori et al. (2017) found that in a certain cell line (A549), ROS could trigger ferroptosis by activating the MAPK pathway. This suggests to us that the MAPK pathway plays an important role not only in apoptosis but also in ferroptosis. p38 MAPK, extracellular signal–regulated kinase 1/2 (ERK1/2), and c-Jun N-terminal kinase (JNK) are the major MAPKs (Chang et al., 2021). In human mammary epithelial cells, the activation of ERK1/2 induces sensitivity to ferroptosis (Poursaitidis et al., 2017). In AML cells, the use of JNK and p38 inhibitors would cause resistance to erastin-induced ferroptosis (Yu et al., 2015). Although MAPK has been proven to be related to the ferroptosis of cancer cells, its contribution to the ferroptosis of ALL cells has not been clearly defined. Clarifying the erastin-triggered interaction between MAPK and ferroptosis is therefore critical for the development of ALL treatment strategies.
In this study, we compared the cell viability; mitochondrial morphological variations; oxidation levels; and System
Materials and Methods
Cell culture
Molt-4 cells, a kind of T cell acute lymphoblastic leukemia (T-ALL) cell line, were incubated in RPMI-1640 (Biological Industries, Israel) containing 10% fetal bovine serum (Biological Industries), 100 U/mL penicillin (Hyclone), and 100 U/mL streptomycin (Hyclone), then cultured in a humid atmosphere containing 5% CO2 at 37°C.
Cell viability measurements
The cells were harvested in the logarithmic growth period (5 × 105), plated into 96-well plates with erastin and various inhibitors, and incubated for 24 h at 37°C and 5% CO2. A total of 10 μL of Cell Counting Kit-8 (Dojindo) reagent was added to each well. The cells were then cultivated for 2–3 h. Subsequently, their optical density values were measured at the absorbance of 450 nm by using a microplate reader (Biotek). Erastin and Ferrostain-1 were purchased from (MedChemExpress), and z-vad-fmk, Nec-1, and 3-MA were procured from Meilunbio (China).
Observation of mitochondria by transmission electron microscopy
Molt-4 cells were seeded into a six-well plate, treated with erastin, and cultured for 24 h. Then, the cells were collected for centrifugation. The cell precipitate was fixed with 1 mL of 2.5% glutaraldehyde at 4°C, washed three times with 0.1 M phosphate buffer, fixed with 1% osmium acid at 4°C, and washed again three times. Subsequently, the cells were dehydrated in an alcohol gradient before being embedded. After curing, the sample was cut into ultrathin sections (Leica, Germany). After the sample was successively stained with uranyl acetate and lead citrate, its mitochondrial ultrastructure was observed with a transmission electron microscope (Hitachi, Japan).
ROS, malondialdehyde, and glutathione assays
The content of ROS in cells was measured using flow cytometry (CytoFLEX S). The cells were harvested, centrifuged for supernatant collection, dyed with 10 μM 2′,7′-dichlorodihydrofluorescein diacetate (DFCH-DA), and incubated in the dark for 30 min. Then, cells were washed with RPMI-1640 two to three times and finally added with 500 μL of phosphate-buffered saline (PBS). Fluorescence intensity of the cells was detected at the excitation wavelength of 488 nm and the emission wavelength of 525 nm. Glutathione (GSH) and malondialdehyde (MDA) contents were measured by using GSH and MDA assay kits from Nanjing Jiancheng Bioengineering Institute (China).
Total RNA extraction, reverse transcription, and quantitative real-time PCR
The total RNA was extracted from erastin-treated Molt-4 cells by using Trizol reagent (Beyotime, China). RNA quantification was performed with a micro ultraviolet–visible spectrophotometer. T100™ Thermal Cycler (Bio-Rad) was used to reverse transcribe 1 μg of total RNA in a 20 μL volume. CFX Connect real-time fluorescence quantitative PCR (qPCR; Bio-Rad) was used for real-time PCR with a 20 μL volume. β-Actin was used as a housekeeping gene. The primer sequences are presented in Table 1. The relative mRNA expression level was calculated using the 2−△△CT method in Excel.
Sequences of the Primers Used for Real Time Quantitative PCR Analysis
Protein extraction and Western blot analysis
Cells were harvested and washed twice with ice-cold PBS then lysed on ice for 15 min with RIPA lysate buffer (Beyotime) containing protease and phosphatase inhibitors. Then, proteins were uniformly mixed with 5 × sodium dodecyl sulfate (SDS) loading buffer and boiled at 100°C for 5 min. Equal amounts of protein (30–40 μg) were loaded and separated by 10% SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and then electrotransferred onto polyvinylidene fluoride (PVDF) membranes. The PVDF membranes were blocked in 5% nonfat milk for 80 min and incubated with p38MAPK, ERK1/2, JNK, p-p38MAPK, p-ERK1/2, p-JNK, and β-actin primary antibodies at 4°C overnight and subsequently incubated with the matching secondary antibodies at room temperature for 2 h. Electrochemiluminescence (ECL) reagent was used to visualize bands. ImageJ software was used to quantify the relative protein contents, and the final results were the target protein/β-actin ratios. The primary antibodies, secondary antibodies, and ECL mentioned above were all from Beyotime.
Statistical analysis
SPSS 22.0 software was used for statistical analysis of data. One-way analysis of variance was performed to compare the difference between multiple groups. Dunnett's t-test was used to analyze the difference between the experimental and control groups. Differences were statistically significant when p < 0.05.
Results
Effect of erastin on Molt-4 cell activity
To test whether erastin can inhibit the ALL cells viability, we treated Molt-4 cells with 0, 2.5, 5, 10, 20, and 40 μM erastin for 12, 24, and 48 h. The results showed that cell viability decreased not only with the increase in erastin concentration, but also with the prolongation of treatment time (Fig. 1). These results revealed that erastin could inhibit the proliferation of Molt-4 cells in a dose- and time-dependent manner.

Changes in the viability of Molt-4 cells treated with different concentrations of erastin for different durations. SPSS 22.0 software calculated that the half-inhibitory concentration of Molt-4 cells treated with erastin for 12, 24, and 48 h were 65.23, 27.68, and 7.69 μM, respectively. Erastin concentrations of 0, 5, 10, and 20 μM were selected for treatment and incubation for 24 h in subsequent experiments. **p < 0.01 (vs. 12 h control group); # p < 0.01 (vs. 24 h control group); & p < 0.01 (vs. 48 h control group); n = 5.
Effects of different cell death inhibitors on cell viability
Early experimental results confirmed that erastin triggered cell death, but whether the mode of death was ferroptosis was still unclear. We treated Molt-4 cells with erastin and inhibitors regulating cell death to further identify the mode of cell death. As given in Figure 2E, cell viability was restored, although not entirely, by the ferroptosis inhibitor Fer-1, indicating that Fer-1 partially reversed the cell death induced by erastin, whereas z-vad-fmk, Nec-1, and 3-MA failed to maintain the viability of Molt-4 cells. The results mentioned above preliminarily indicated that the erastin-induced death pathway of Molt-4 cells was ferroptosis rather than apoptosis, necroptosis, or autophagy.
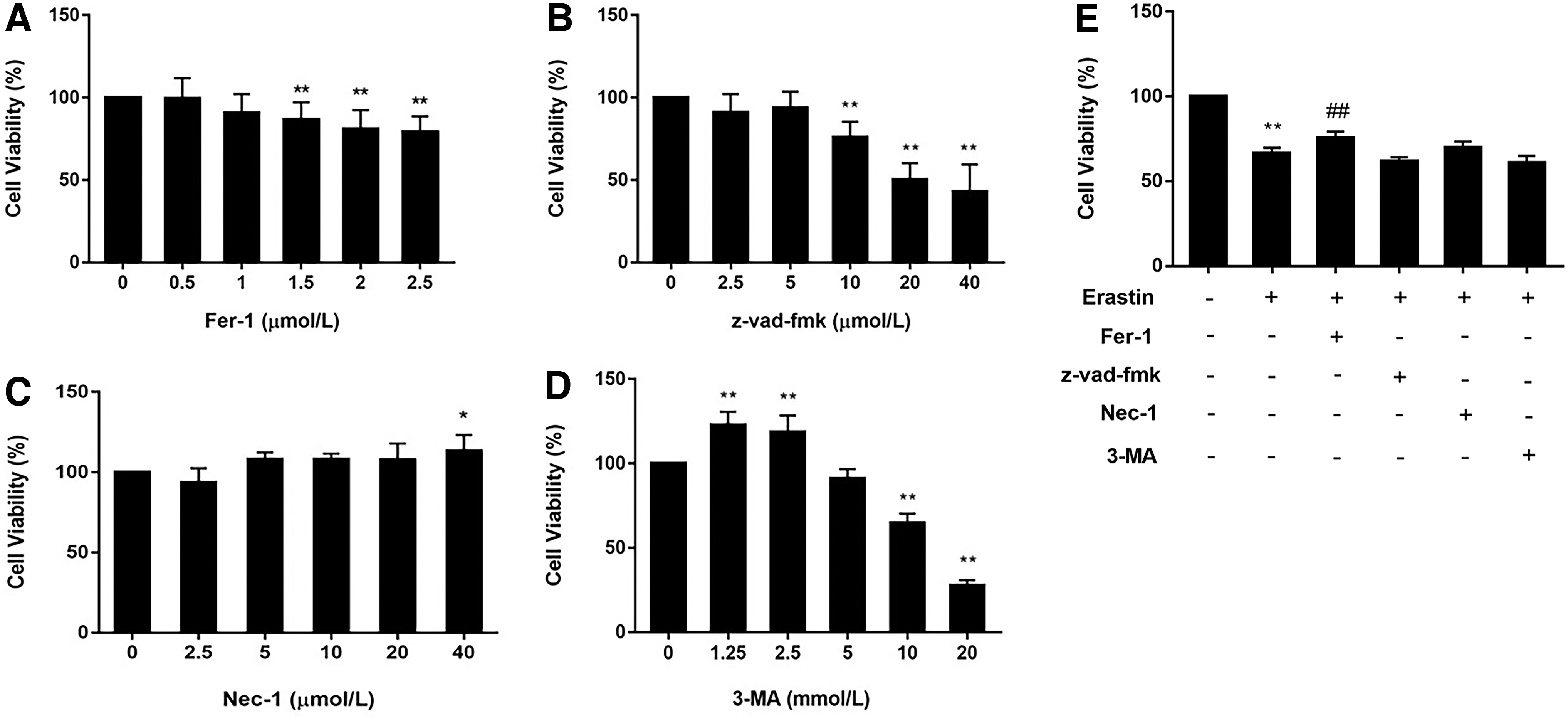
Changes in cell viability after treatment with different cell death inhibitors and erastin.
Ultrastructure of mitochondria in erastin-treated Molt-4 cells
The above experiments have preliminarily provided that erastin-induced ferroptosis is distinct from other forms of death. However, the dramatic morphological changes of mitochondria in ferroptosis are convincing proof to distinguish ferroptosis from other forms of cell death. The typical morphological changes associated with ferroptosis include volume reduction in mitochondria, reduction or disappearance of mitochondrial cristae, and increase in bilayer membrane density (Dixon et al., 2012).
As given in Figure 3A, a, the mitochondria of untreated Molt-4 cells had a regular shape, size, complete structure, and orderly cristae. Erastin-treated cells exhibit increased mitochondrial membrane density and corresponding volume reduction (Fig. 3B–D, b–d). These changes in erastin-treated Molt-4 cells were the same as the characteristics of ferroptosis, and no characteristic morphological features associated with necroptosis (swelling of cytoplasm, rupture of plasma membrane), apoptosis (condensation and margination of chromatin), or autophagy (formation of double membrane–enclosed vesicles) (Dehart et al., 2018). The mode of erastin-induced death is preliminarily shown to be ferroptosis.

Ultrastructure of mitochondria in Molt-4 cells under transmission electron microscopy. The red box in
Erastin inhibits antioxidant defense in Molt-4 cells
The dominant feature of ferroptosis is the lethal accumulation of lipid peroxides and ROS (Li and Li, 2020). In our study, we used DCFH-DA assay to measure the level of ROS. This assay is based on the diffusion of DCFH-DA into the cells. Then it is deacetylated by cellular esterases to a nonfluorescent compound, which is later oxidized by ROS into DCF. DCF is highly fluorescent and its fluorescence intensity could represent the level of ROS in cells. In Figure 4A, as the concentration of erastin was increased, the ROS peak curve shifted to the right, and the 2′,7′-dichlorofluorescein (DFC) fluorescence intensity of Molt-4 cells gradually increased (Fig. 4B).
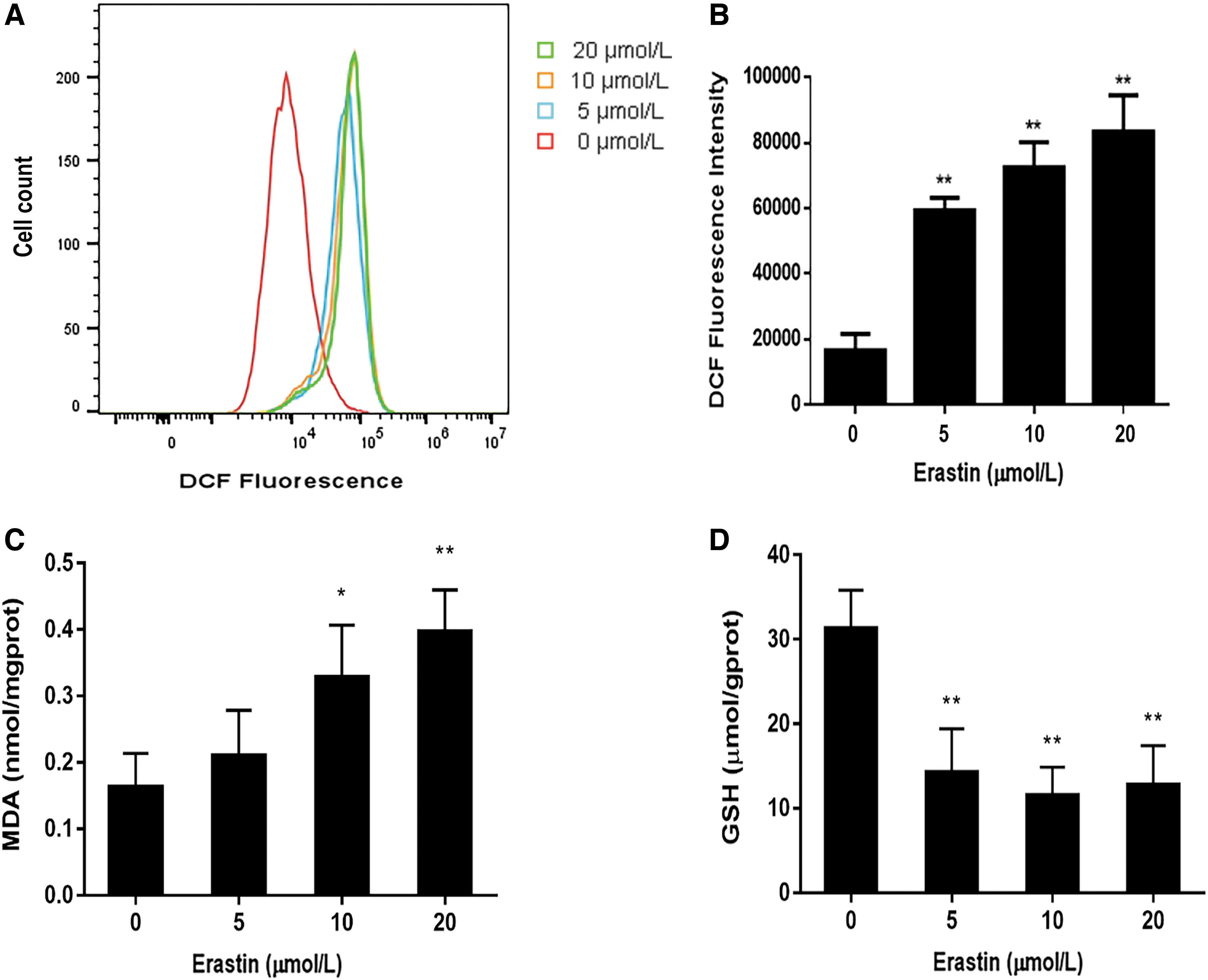
Lipid peroxidation levels of Molt-4 cells under erastin treatment.
MDA is the main product of lipid peroxidation reactions and is indicative of the level of intracellular peroxides to some extent. In accordance with the levels of ROS, the MDA levels in the 10 and 20 μM erastin-treated groups showed a significant increase (Fig. 4C), indicating that erastin induced lipid peroxidation. The level of GSH, an important intracellular antioxidant, reflects the capacity of an organism to clear ROS (Yang et al., 2014). Erastin caused a dose-dependent decrease in GSH levels after 24 h of exposure (Fig. 4D). The suppression of the antioxidant system and the elevation of oxidative damage further confirmed that erastin triggered ferroptosis in Molt-4 cells.
Regulation of System
, GPX4, and MAPK in erastin-triggered ferroptosis
It has been suggested that both inhibition of System
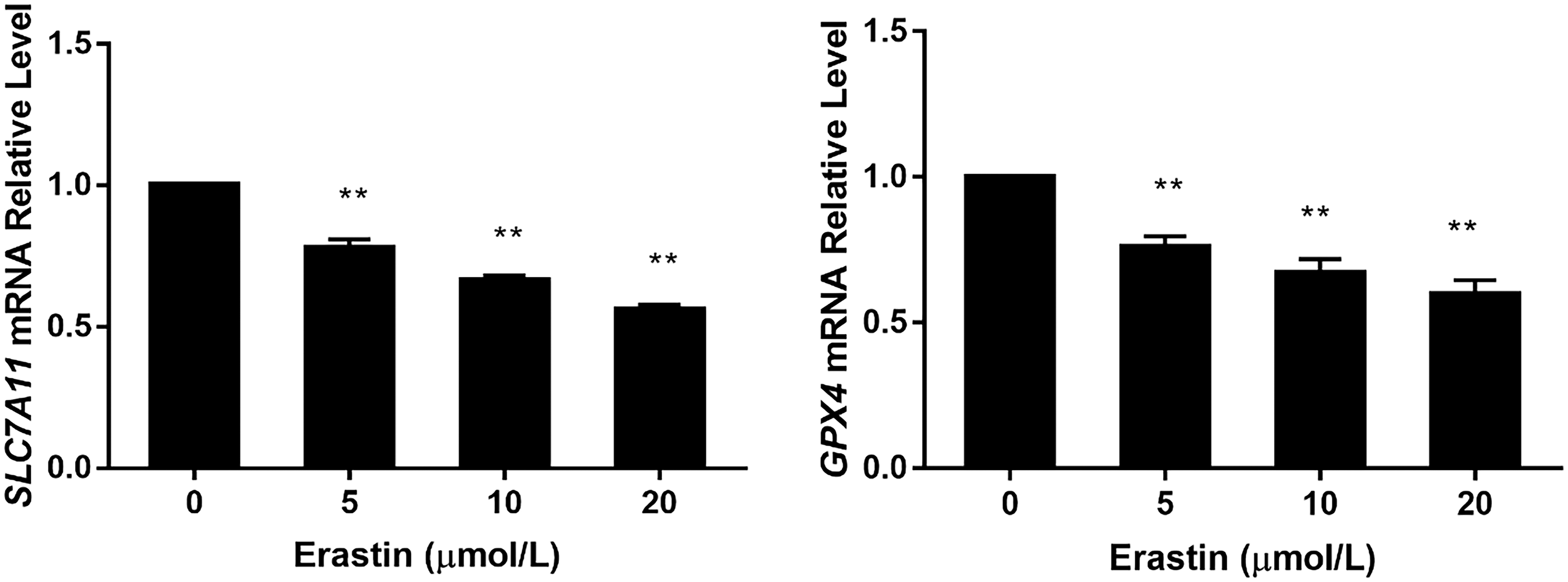
Expression levels of SLC7A11 and GPX4 mRNA after erastin treatment; **p < 0.01 (vs. control group); n = 4. GPX4, glutathione peroxidase 4.
The results mentioned above showed that erastin inhibited the action of System

Expression levels of p38 MAPK, ERK1/2, and JNK and their phosphorylated proteins in Molt-4 cells under erastin treatment.
The cells were treated with p38 MAPK, ERK1/2, and JNK inhibitors to further elucidate the effects of p38 MAPK, ERK1/2, and JNK. Cell viability assays showed that in Molt-4 cells, the inhibition of p38 MAPK rescued erastin-induced ferroptosis. By contrast, ERK1/2 inhibitors exacerbated erastin-induced ferroptosis (Fig. 6H). These findings, combined with the results of Western blot analysis, indicated that p38 MAPK is involved in erastin-induced ferroptosis.
Discussion
A growing number of studies have shown that ferroptosis is associated with tumor suppression (Jiang et al., 2015; Viswanathan et al., 2017; Tang et al., 2021). Nevertheless, the application of ferroptosis induction in hematological malignancies is limited. Treatment with a low dose of erastin in combination with cytarabine and adriamycin results in a more favorable prognosis for AML than conventional chemotherapy alone (Yu et al., 2017). However, little is known about ALL. In the present study, we found that Molt-4 cells are sensitive to erastin-induced ferroptosis and that this process also involves the inhibition of System
Ferroptosis is a form of cell death that is different from autophagy, apoptosis, and necroptosis (Dixon et al., 2012). It can be triggered in cancer cell lines by a number of small molecules, including (1S,3R)-RSL3 and erastin (Dachert et al., 2016). Recent researches have shown that erastin triggered ferroptosis in human hepatocellular cells and human promyelocytic leukemia cells (HL60) (Yuan et al., 2016; Ye et al., 2019). To investigate the effect of erastin on ALL cells, we treated Molt-4 cells with different concentrations of erastin. Unrestrained lipid peroxidation is the hallmark of ferroptosis (Li and Li, 2020; Jiang et al., 2021); we detected the oxidative damage-related markers. The levels of ROS and MDA were increased.
Alterations of mitochondrial, condensed mitochondrial membrane densities and smaller volume, as well as the diminished or vanished of mitochondria crista and outer membrane ruptured, were consistent with the ferroptosis characteristics (Dixon et al., 2012; Li et al., 2021). Furthermore, only Fer-1 restored cell viability after treatment with the Fer-1, z-vad-fmk, Nec-1, and 3-MA in combination with erastin. These findings confirmed that erastin has triggered ferroptosis in Molt-4 cells, which is consistent with previous studies (Dachert et al., 2016; Pontel et al., 2022).
Ferroptosis is regulated by multiple mechanisms, the model of action of erastin-triggered ferroptosis is distinct from that of RSL3 that directly inhibit GPX4 (Yang et al., 2014). It depletes GSH by preventing cystine uptake by regulating System
Studies have proved that erastin-induced ferroptosis is closely associated with the accumulation of ROS (Yagoda et al., 2007; Dixon et al., 2012). ROS of erastin-treated Molt-4 cells is significantly increased. It has been suggested MAPK is tightly linked to ferroptosis in multiple types of diseases (Gao et al., 2018). Li et al. (2018) found that the inhibition of p38 MAPK activation in germ cells with high ROS prevents ferroptosis. Codenotti et al. (2018) suggested that erastin triggers ferroptosis in highly proliferating myogenic lines through ERK pathway-dependent manner. Along with the ferroptosis feature of ROS accumulation in Molt-4 cells, we further explored the role of MAPK in erastin-induced ferroptosis. Erastin promoted p38 MAPK, ERK1/2, and JNK protein expression and phosphorylation of p38 MAPK, ERK1/2, and JNK in Molt-4 cells. The cell viability results showed that p38 MAPK inhibitors alleviated erastin-induced ferroptosis, in agreement with previous findings (Yu et al., 2015; Hattori et al., 2017).
It is notable that although ERK1/2 was activated, the inhibitor of ERK1/2 exacerbated erastin-induced ferroptosis. This is not consistent with previous studies (Codenotti et al., 2018; Gao et al., 2018). After intensive investigation, we speculated that, first, 50 μM of ERK1/2 inhibitor does not affect other cells (de Rooij et al., 2022), but it may reduce the metabolic ability of Molt-4 cells or trigger other forms of cell death. Second, Su et al. (2019) reported that panx1 knockdown inhibits erastin-induced ferroptosis by activating the MAPK/ERK pathway, and this protective effect is associated with increased expression of antioxidant gene HO-1.
It might be thought, in Molt-4 cells, p38 MAPK and ERK1/2 simultaneously regulate erastin-induced ferroptosis. But the positive regulation of p38 MAPK has a greater effect than the negative regulation of ERK1/2. That would explain why the cell viability is diminished after being treated with ERK1/2 inhibition, although ERK1/2 is activated when Molt-4 cells undergo ferroptosis. This controversy deserves further thorough investigation and exploration. In consideration of the above results, we suggest that the activation of p38 MAPK and ERK1/2 is associated with erastin-induced ferroptosis in Molt-4 cells. The specific mechanism involving the upstream and downstream molecules of the p38 MAPK and ERK1/2 pathway in the regulation of ferroptosis remains unclear and will be further explored in well-designed experiments in the future.
Conclusions
The findings of this study included confirmation of the role of erastin in inducing ferroptosis in T-ALL cells and a preliminary investigation of the putative mechanisms regulating ferroptosis via the MAPK pathway (Fig. 7). System

Schematic of proposed mechanisms for erastin regulating ferroptosis in Molt-4 cells. When Molt-4 cells are exposed to erastin, the System
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the Natural Science Foundation of Hubei Province (2019CFB500).