
Abstract
Folate, as the initial substrate in one-carbon metabolism, is involved in the synthesis of important substances such as DNA, RNA, and protein. Folate deficiency (FD) is associated with male subfertility and impaired spermatogenesis, yet the underlying mechanisms are poorly understood. In the present study, we established an animal model of FD to investigate the effect of FD on spermatogenesis. GC-1 spermatogonia were used as a model to investigate the effect of FD on proliferation, viability, and chromosomal instability (CIN). Furthermore, we explored the expression of core genes and proteins of spindle assembly checkpoint (SAC), a signaling cascade ensuring accurate chromosome segregation and preventing CIN during mitosis. Cells were maintained in medium containing 0, 20, 200, or 2000 nM folate for 14 days. CIN was measured by using a cytokinesis-blocked micronucleus cytome assay. We found that sperm counts decreased significantly (p < 0.001) and the rate of sperm with defects in the head increased significantly (p < 0.05) in FD diet mice. We also found, relative to the folate-sufficient conditions (2000 nM), cells cultured with 0, 20, or 200 nM folate exhibited delayed growth and increased apoptosis in an inverse dose-dependent manner. FD (0, 20, or 200 nM) significantly induced CIN (p < 0.001, p < 0.001, and p < 0.05, respectively). Moreover, FD significantly and inverse dose dependently increased the mRNA and protein expression of several key SAC-related genes. The results indicate that FD impairs SAC activity, which contributes to mitotic aberrations and CIN. These findings establish a novel association between FD and SAC dysfunction. Thus, FD-impaired spermatogenesis may be partly due to genomic instability and proliferation inhibition of spermatogonia.
Introduction
As an important acceptor and donor of a one-carbon unit of various oxidation levels in living organisms, folate is involved in the synthesis of purine and thymine, amino acid metabolism, and DNA methylation (Coppedè, 2015; Fox and Stover, 2008; Liu et al., 2020). Considering the essential functions of folate in biosynthesis and methylation, a growing number of studies have focused on the relationship between folate deficiency (FD) and oncogenesis and reproductive health. Accumulated evidence has shown that FD is associated with male subfertility and impaired spermatogenesis (Huang et al., 2020; Lambrot et al., 2013; Lee, 2021; Lismer et al., 2021; Swayne et al., 2012; Wang et al., 2022; Wong et al., 2000; Yuan et al., 2017). However, the mechanism by which FD impairs spermatogenesis is complex and remains unclear.
Chromosomal instability (CIN) that occurs in germ cells during mitosis and meiosis may lead to sperm DNA damage and aneuploidy, which cause male subfertility and increased risk of chromosomal anomalies in the offspring. It has been established that low folate in seminal plasma is detrimental to sperm DNA stability (Boxmeer et al., 2009; Duthie et al., 2002; Wang et al., 2022). Men with high folate intake have lower overall frequencies of several types of aneuploid sperm such as X disomy, 21 disomy, and sex chromosome nullisomy (Young et al., 2008). This evidence indicates that folate plays a key role in maintaining genomic stability. However, safe concentrations of folate for germ cells to maintain genomic stability have not been reported because most germ cells are in a state of rapid proliferation.
Despite the theoretical and experimental efforts made during the last decades in evaluating the relationship between FD and CIN, the mechanisms underlying FD-induced CIN have not yet been elucidated. Based on previous studies, FD increases chromosome breaks and uracil misincorporation during DNA replication (Blount et al., 1997; Linhart et al., 2009). However, it has been estimated that only a small proportion of micronucleus (MN, a measure of CIN) originates from broken chromosomes (Fenech et al., 2011), suggesting that there are other causes that contribute to the increase in FD-induced CIN. Spindle assembly checkpoint (SAC) is a signaling cascade that ensures accurate chromosome segregation by delaying anaphase initiation until all chromosomes are properly attached to the mitotic spindle. There has been increasing evidence that SAC dysfunction could lead to mitotic aberrations and CIN (Guo et al., 2017; Shimoi et al., 2019; Silva et al., 2018; Uchida et al., 2021; Yu et al., 2022; Yu et al., 2016).
This eventuality raises another question: does FD impair the function of SAC? As mentioned above, folate is an important donor of a one-carbon unit. Deficiency of folate leads to DNA hypomethylation in genomic DNA. It has been shown that folate regulates gene expression by altering the methylation status of gene promoter regions. So far, it is not clear whether the expression of SAC-related genes is regulated by DNA methylation and whether FD regulated the expression of SAC-related genes by promoter demethylation. In this study, we investigated an effect of 5-Azacytidine (5-AZA), a DNA methyltransferase inhibitor, on the expression of SAC-related genes to predict the sensitivity of the expression of these genes to promoter methylation regulation. The present study will provide a basis for further study on the mechanism of SAC gene expression changes caused by FD.
Materials and Methods
Experimental animal modeling for FD
The Animal model of FD was established by referring to the guidelines in the study of Lambrot et al. (2013). Inbred C57BL/6 mice were purchased from Skbes Biotechnology Co., LTD. (Henan, China). To generate experimental males, female C57BL/6 were fed the FD diet (0.3 mg folic acid per kilogram, n = 12; Beijing HFK Bioscience Co. Ltd.). The control group (n = 12) and nonexperimental C57BL/6 males (n = 12) (used for breeding to generate experimental males) were fed regular mouse chow. Females were maintained on the experimental diets through pregnancy and lactation. From weaning at about postnatal day 21, male offspring were given the same experimental diet as their in utero exposure until killing as adults. All mice were housed in vivarium cages under standard conditions (25°C a controlled light/dark cycle). This research received ethics approval from the Ethics Committee of Bengbu Medical College ([2022] No. 144).
Sperm counts and histopathological assays
Experimental males in the FD group and control group were sacrificed for cervical dislocation at postnatal days 40 (control, n = 5; FD, n = 5) and 70 (control, n = 9; FD, n = 10). Each experiment was carried out at least three times. The two caput epididymides were used for each animal at postnatal days 70 for sperm counting and Periodic Acid-Schiff (PAS) staining. Epididymis was collected from each group and homogenized in 1 mL of phosphate-buffered saline (PBS). The homogenized samples were incubated in a 95% humidity and 5% carbon dioxide (CO2) atmosphere at 37°C for 30 min. Epididymal sperm count was routinely determined using a hemacytometer. PAS staining was used as assessment of sperm morphology. To evaluate rate of sperm deformation, abnormal sperm types, such as “hookless,” “banana,” “amorphous,” and “folded” sperms, were counted.
The testis and epididymis samples were fixed overnight in 4% paraformaldehyde solution and embedded in paraffin. Sectionalized slices were dewaxed three times in d-Limonene solution and 100% ethanol solution for 10 min each, followed by gradient dewaxing in ethanol (90%, 80%, and 50%) for 2 min and Milliq-H2O for 5 min. The histological changes were observed by Hematoxylin and Eosin (H&E) staining under a light microscope.
Cell culture and treatment with folate
GC-1spg (ATCC® CRL-2053™), a cell line derived from Mus musculus that shows characteristics of a stage between type B spermatogonia and primary spermatocytes, was obtained from Procell Life Science & Technology Co., Ltd. (Wuhan, China). Cells were cultured in standard Dulbecco's modified Eagle's medium (DMEM, high glucose) (BI, Kibbutz Beit Haemek, Israel) supplemented with 5% heat-inactivated fetal bovine serum (Gibco BRL, Grand Island, NY, USA), 0.1% penicillin (5000 IU/mL)/streptomycin (5 mg/mL) solution (Gibco), and 1% l-glutamine (2 mM; Sigma-Aldrich, St. Louis, MO, USA). Cells were kept at 37°C in a humidified atmosphere of 5% CO2. Then, cells were cultured in folate-free DMEM (BI) for folate depletion. Folate (folic acid; Sigma-Aldrich) was added to this medium at a final concentration of 20, 200, or 2000 nM. A steady state was achieved after 11 days of continuous FD culture. These concentrations have been used and described previously (Francois et al., 2016). The absence of folate (0 nM) was selected as a positive control for severe FD.
The concentration of 20 nM folate was selected for low folate because, based on several in vitro studies, it causes genetic damage. The concentration of 2000 nM folate was selected as representative of a folate-sufficient (FS) condition because it is the folate concentration found in most standard culture media.
Cell proliferation, viability, and cell apoptosis assays
To confirm the proliferation rate of GC-1 cell line, cell growth curve was determined by cell counting. Cells were transferred into prewarmed FD and FS complete medium before experiments. Total cell counts using a hemocytometer and viability estimates using Trypan Blue were performed for running 6 days. To determine the cell viability, GC-1 cells cultured with various folate concentrations were maintained for 11 days in T-25 flasks. Subsequently, cells were seeded in 96-well plates at a density of 3 × 103 cells/well and cultured overnight. On days 12, 13, and 14, cell viability was estimated by using the Cell Counting Kit-8 colorimetric assay (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) according to the manufacturer's protocol. At the same time, cultures were maintained for 14 days for the cell apoptosis assay. The proportions of apoptotic cells were evaluated by using the Annexin V/Fluorescein Isothiocyanate Staining Kit (Beyotime, Shanghai, China) according to the manufacturer's protocol. All cells were analyzed by using a flow cytometer (BD Accuri™ C6 1.0.264.21, BD Biosciences, San Jose, CA, USA).
Treatment of GC-1 cells with 5-AZA
The 5-AZA is a DNA and RNA nucleoside cytidine analog that is typically used to activate the expression of genes by promoter demethylation. To investigate epigenetic regulation of SAC-related genes, GC-1 cells were treated with different concentrations of 5-AZA (Sigma-Aldrich). In brief, GC-1 cells were cultured in standard DMEM. Subsequently, cells were seeded in six-well plates at a density of 1 × 106 cells/well. After 24 h of culture, the whole culture medium was removed from each well and cells were washed with PBS once. Then cells were cultured in DMEM containing different concentrations of 5-AZA (1, 10, or 50 μM) for 48 h.
Real-time quantitative polymerase chain reaction
Total RNA was extracted from GC-1 cells using TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and was utilized to synthesize complementary DNA (cDNA) with the PrimeScript™ RT Reagent Kit (TaKaRa Bio, Inc., Otsu, Japan) according to the manufacturer's protocol. Real-time quantitative polymerase chain reaction (RT-qPCR) was performed in triplicate by using the SYBR® Premix Ex Taq™ II Kit (TaKaRa Bio, Inc.) on an Applied Biosystems Quant Studio™ 6 Flex thermocycler (Thermo Fisher Scientific, Inc.). The expression of SAC-related genes, including Bub1, Bub3, BubR1, Mad1, Mad2, and Cdc20, was analyzed. Meanwhile, expression of the apoptosis-related genes Casp3 and Bcl2 was analyzed to confirm the flow cytometry results. The primer sequences for the tested genes are presented in Table 1. The thermal cycling conditions were 95°C for 5 min, followed by 40 cycles of 95°C for 5 s and 60°C for 34 s. Gene expression was normalized to that of housekeeping gene Gapdh. The fold-change for each mRNA was calculated by using the 2–ΔΔCt method.
Primers for Real-Time Quantitative Polymerase Chain Reaction
Cytokinesis-block micronucleus cytome assay
CIN in GC-1 cells cultured with different folate concentrations was measured by using the previously described cytokinesis-block micronucleus cytome (CBMN-Cyt) assay (Bull et al., 2012; Thomas and Fenech, 2011), with some modifications based on the findings of our pilot experiment (significant proliferative and morphological change of GC-1 cells can be observed after 12 days of culture in FD). In brief, GC-1 cells were cultured with various folate concentrations (0, 20, 200, or 2000 nM) for 12 days in T-25 flasks. Subsequently, cells were seeded in 24-well plates at a density of 5 × 105 cells/well. Following a 24-h incubation at 37°C in 5% CO2, cytochalasin B (4.5 μg/mL; Sigma-Aldrich) was added to block cytokinesis, and cells were harvested after another 28-h by cytocentrifugation using a cytocentrifuge (Xiangyi Centrifuge Instrument Co., Ltd., Changsha, China).
Cell suspensions were centrifuged at 800 rpm for 6 min at 25°C. Then the supernatant was removed. The cell pellet was used to prepare a cellular smear. Slides were then air-dried, fixed with methanol–glacial acetic acid (3:1, v/v), air dried again, fixed in fresh methanol–glacial acetic acid (3:1, v/v) for 15 min and stained with 5% Giemsa.
Binucleated cells (BNCs) and major genetic damage markers (MN, nuclear plasma bridge [NPB], and nuclear buds [NBuds]) were examined with an optical microscope (1000 × total magnification; Olympus, Tokyo, Japan) and scored based on criteria described by Fenech et al. (2011). One thousand BNCs were scored per replicate and at least 3000 BNCs were analyzed for each folate concentration.
Western blot analysis
Following treatments, protein was extracted from GC-1 cells by using radioimmunoprecipitation assay lysis buffer with 1% protease inhibitor cocktail and 1 mM phenylmethylsulphonyl fluoride (Beyotime). Protein concentrations were determined by using the Bicinchoninic Acid (BCA) Assay Kit (Beyotime). Equivalent amounts of protein from each sample were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred to polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA). The membranes were subjected to immunoblotting with primary antibodies against Mad2, Bub1, Bub3, BubR1 (Bethyl Laboratories, Montgomery, TX, USA) or β-actin (Cell Signaling Technology, Danvers, MA, USA). The blots were visualized by using a Bio-Rad ChemiDoc XRS imaging system (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analyses
Each experiment was carried out at least three times and the data are presented as the mean ± standard deviation. The data assessing serum folate concentration, sperm counts, and abnormal sperm rate were analyzed for differences between the control and treated group by unpaired Student's t-test or in case of failed normality by the Mann–Whitney U-test. The differences in the frequency of BNCs with CIN biomarkers and relative mRNA expression level of genes were analyzed by using one-way analysis of variance, and the Q test was used to determine differences between groups. A p < 0.05 (two-tailed test) was considered to be statistically significant. All statistical analyses were performed using SPSS Statistics version 17.0 (SPSS, Inc., Chicago, IL, USA).
Results
Effects of FD on sperm quality and testicular structure
To verify whether the animal model was successfully constructed, the concentration of mouse plasma folic acid was determined by folic acid enzyme-linked immunosorbent assay. The total plasma folate levels in the FD group (n = 4) were significantly lower than those in the control group (n = 4, p < 0.05; Fig. 1A). Compared with the control group, the FD diet did not affect the testicular organ index (data not shown). However, histological examination of adult testes (10 weeks) revealed significant morphological differences between control and FD mice. Conventional epididymal semen analysis showed that the sperm count of the FD group (n = 5) was significantly lower compared with the control group (n = 4, p < 0.001; Fig. 1B). Furthermore, careful examination of the sperm morphology by PAS staining showed that FD mice produced higher numbers of sperm with defects in the head, suggesting that FD possibly caused a developmental defect in the maturation of the sperm head (control, n = 3, FD, n = 3, p < 0.05; Fig. 1C, E). The pathological changes in testicular tissues were observed by H&E staining.
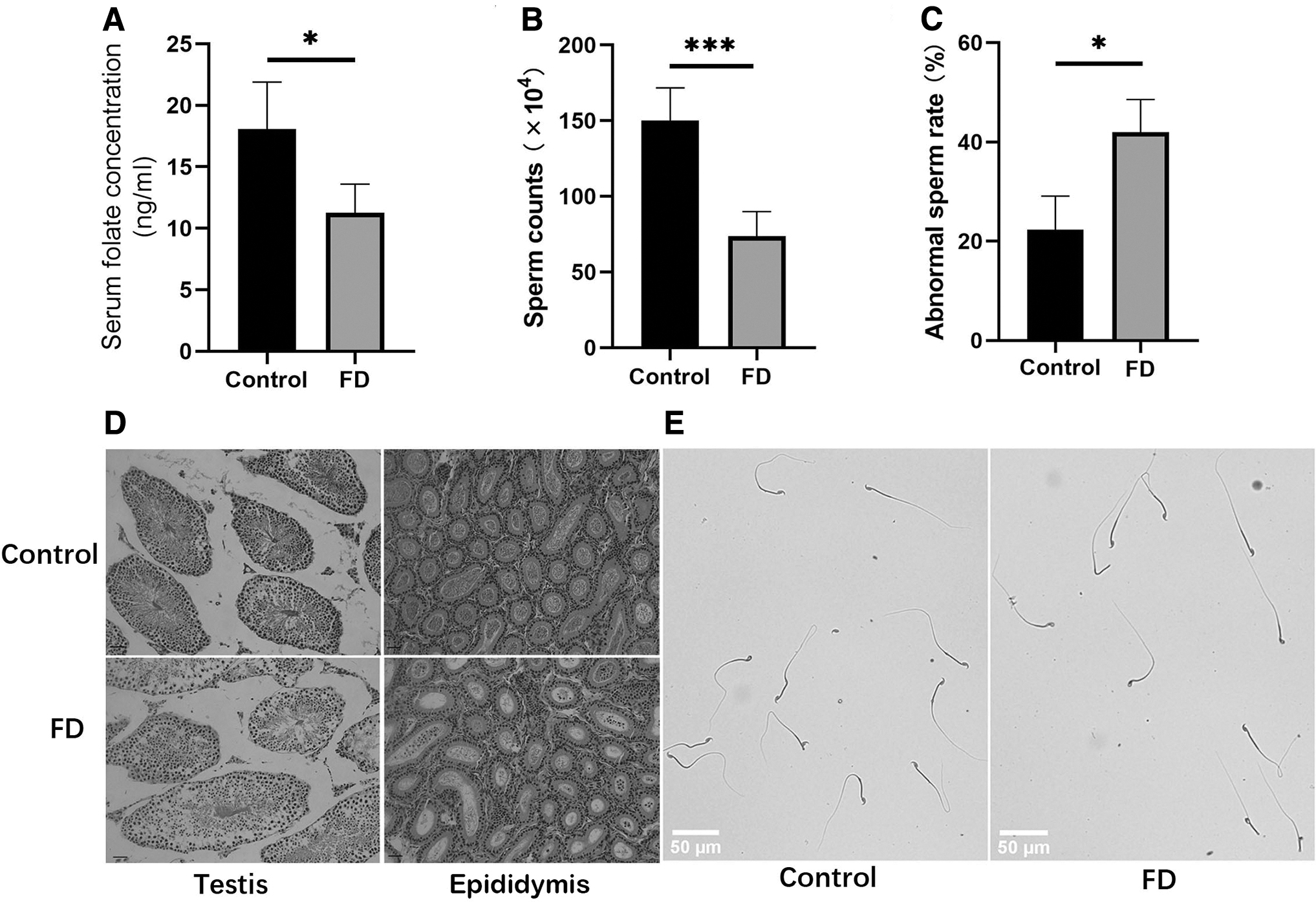
Effects of FD on sperm quality and testicular structure.
Irregular seminiferous tubules were observed with large space around them and a reduced number of germ cells in the FD group (Fig. 1D). In addition, compared with the control group, the number of sperm in the epididymal duct was reduced significantly in the FD group (Fig. 1D). In Figure 2, different types of germ cells were indicated by the arrow in testicular section at postnatal days 40. The results showed that germ cells were denser and neatly arranged in control group, but looser and irregularly arranged in FD group. Moreover, spermatogonia residing on the basement membrane are very tightly packed in control group but loosely packed in FD group.
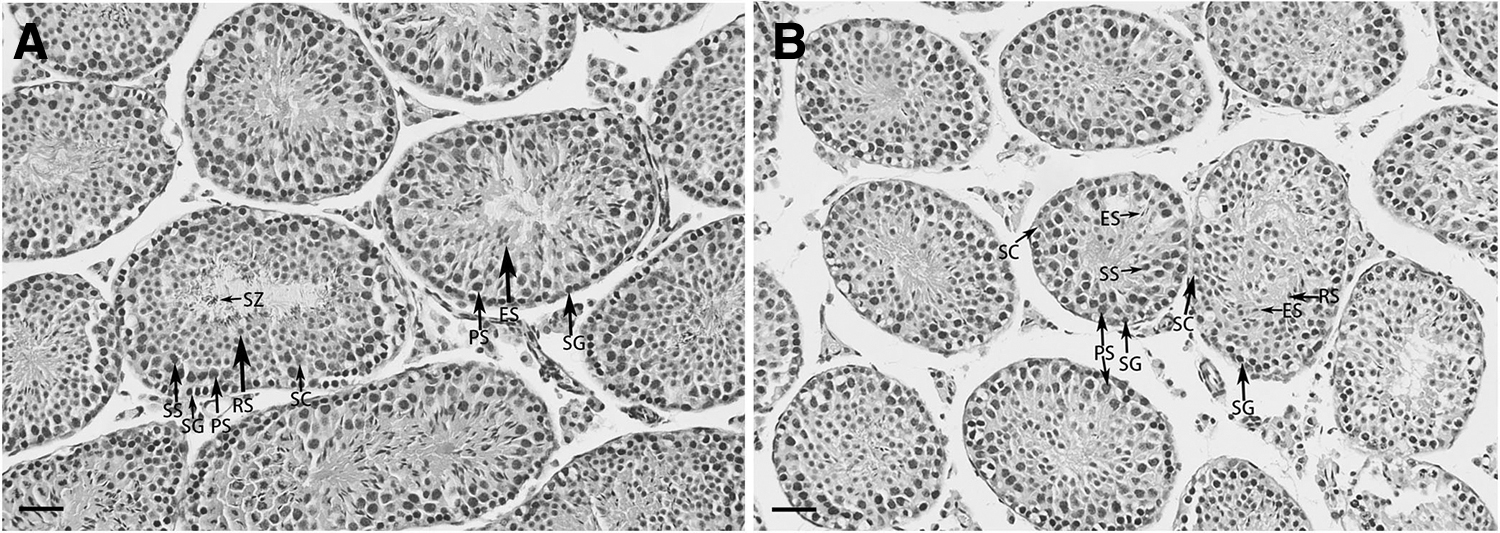
Histological examination of testis at postnatal day 40.
FD inhibited proliferation, viability, and enhanced apoptosis of GC-1 cells
As the incubation time increased, the growth rate of GC-1 cells gradually decreased, except for the FS culture. After entering the logarithmic growth phase, the cells proliferated rapidly in FS culture (Fig. 3B). GC-1 cells have a population doubling time of about 24 h. The result indicated that GC-1 cells have a fast proliferation rate in vitro. However, the cells cultured with 20 nM folate showed significant growth delay (Fig. 3B). After 2 weeks, the cells cultured with 0 or 20 nM folate showed abnormal morphology, such as increased size and a flattened and diffuse cell outline, after which the cells almost stopped growing (Fig. 3A). However, similarly to the FS culture, cells cultured with 200 nM folate showed normal morphology. Cells cultured with 0, 20, or 200 nM folate showed a significant, dose-dependent decrease in viability (p < 0.001; Fig. 3C). The results of proliferation kinetics showed that FD significantly delayed the growth of GC-1 cells.
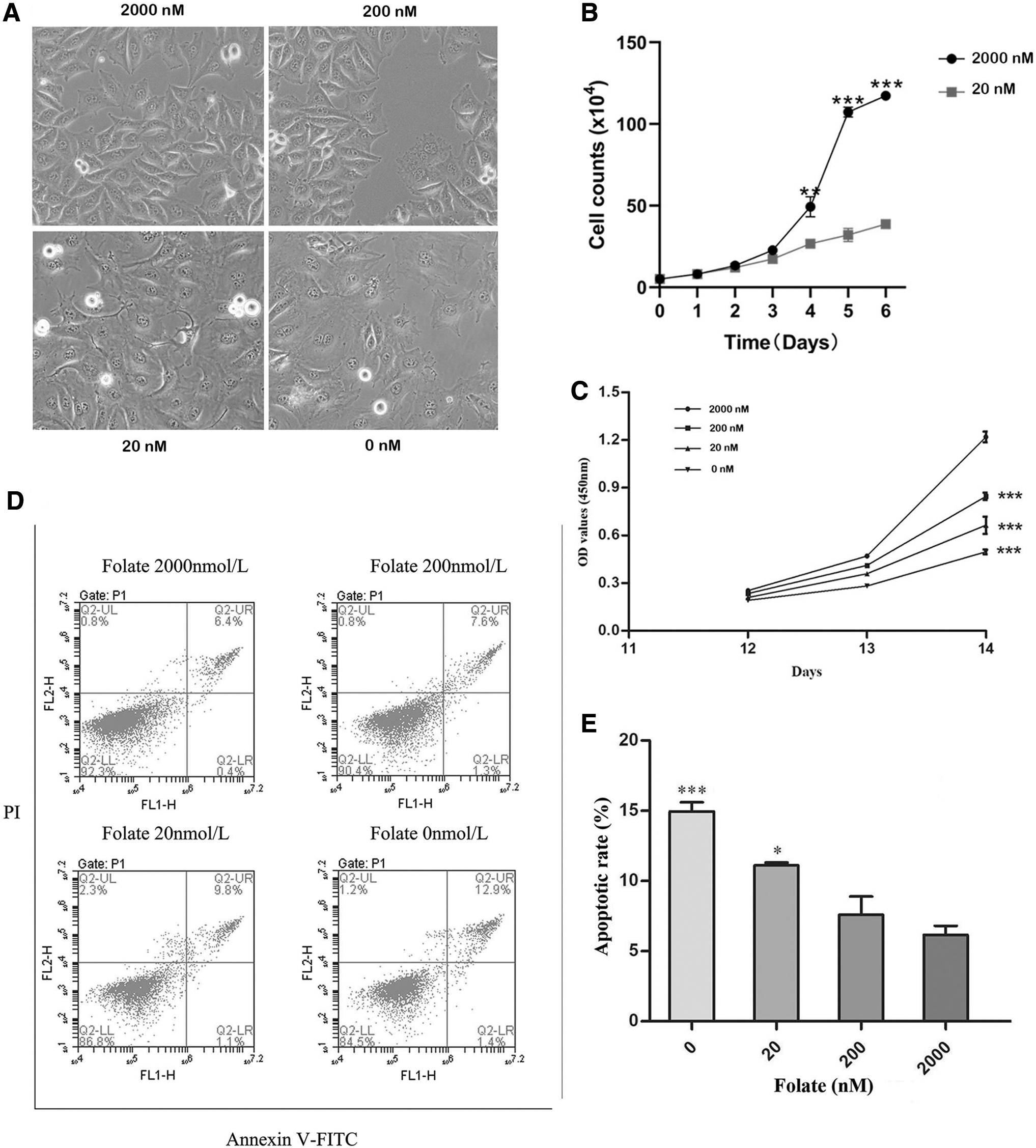
FD inhibits proliferation and enhances apoptosis. GC-1 cells were cultured with various folate concentrations for 14 days.
There were also significant differences in apoptosis and cell death among the various groups after 14 days of culture. As presented in Figure 3D, E compared with the FS culture, cells cultured with 0 or 20 nM folate showed a significant, inverse dose-dependent increase in apoptosis (p < 0.001 and p < 0.05, respectively). Although the apoptotic rate of the cells treated with 200 nM folate was higher compared with the FS culture, the difference was not significant (p > 0.05). Hence, the decline in cell viability in FD was due to a marked increase in apoptosis and cell death.
FD induced cytogenetic damage in GC-1 cells
After 2 weeks of culture, the frequency of BNCs with one or more of the CIN biomarkers (MN, NPB, and NBuds) increased significantly as the folate concentration decreased. Compared with the FS culture, there was an increased frequency of total genetic damage (MN and/or NPB and/or NBud) in cells cultured with 0, 20, or 200 nM folate (p < 0.001, p < 0.001, and p < 0.05, respectively; Fig. 4A, B). Compared with the FS culture, the frequency of BNCs with MN (the main CIN biomarker) at day 14 increased 9.9-fold in cells cultured with 0 nM folate, 3.1-fold in cells cultured with 20 nM folate, and 1.9-fold in cells cultured with 200 nM folate (Fig. 4C). Meanwhile, compared with the FS culture, the frequency of BNCs with NBuds increased 6.2-fold in cells cultured with 0 nM folate and 2.5-fold in cells cultured with 20 nM folate (Fig. 4D). However, the difference between the FS culture and cells cultured with 200 nM folate was not significant (p > 0.05). These data indicate that the folate concentration is correlated with genomic stability, and 200 nM folate is insufficient to protect the genomic stability of GC-1 cells in vitro.

FD induced CIN in GC-1 cells. Cells were cultured with various folate concentrations for 14 days.
FD upregulated the expression of key SAC-related genes
To test our hypothesis that FD impairs SAC, which may be the mechanism behind FD-induced CIN, we investigated the expression of several key SAC-related genes, including Mad1, Mad2, Bub1, Bub3, BubR1, and Cdc20 (Fig. 5A). Increasing evidence has shown that altered expression of one or more of these genes leads to SAC dysfunction. FD significantly and inverse dose dependently increased the expression of some of SAC-related genes. Specifically, Mad2, Bub1, and BubR1 were upregulated significantly in cells cultured with 0 or 20 nM (p < 0.01) compared with the FS culture.

Effect of FD on the relative expression of key SAC-related genes in GC-1 cells.
For example, the expression of Mad2 was increased over seven-fold in cells cultured with 0 nM folate (p < 0.01) and almost five-fold in cells cultured with 20 nM folate (p < 0.01). The expression of Bub1 was increased over 15-fold in cells cultured with 0 nM folate (p < 0.001) and over 10-fold in cells cultured with 20 nM folate (p < 0.001). However, there was only a slight increase in expression of Bub3 (p < 0.01). In addition, the expression of Mad1 and Cdc20 were increased significantly in cells cultured with 0 nM folate (p < 0.001) or 20 nM folate (p < 0.001 and p < 0.05, respectively).
Both genes were not upregulated significantly in cells cultured with 200 nM folate (p > 0.05). To further confirm the results of RT-qPCR, we used western blotting to examine the protein expression of Mad2, Bub1, Bub3, and BubR1. As shown in Figure 5C, FD upregulated the core protein expression of SAC, except for Bub3. The protein expression basically coincide with the gene expression results (Fig. 5B,C).
We also found FD significantly and inverse dose dependently increased the expression of the oncogenic transcription factor c-MYC gene (Fig. 5D). Furthermore, to test the potential involvement of DNA methylation in the regulation of SAC-related gene expression, we treated GC-1 cells with different concentrations of 5-AZA, a DNA methylation inhibitor. The results showed that the expression of Mad2, Bub1, and BubR1 was increased significantly and dose dependently (Fig. 5E).
Discussion
Although studies have suggested an association between folate status and spermatogenesis, the mechanisms underlying FD-induced oligozoospermia remain unclear (Ly et al., 2017; Swayne et al., 2012). In the present study, we established an animal model and a GC-1 spermatogonia cell model of FD to investigate the effect of FD on spermatogenesis. FD impaired spermatogenesis in vivo. FD inhibited GC-1 proliferation and viability and led to CIN in vitro. Moreover, FD deregulated the expression of core SAC-related genes. Our findings suggest that SAC dysfunction induced by FD may play a role in mitotic aberrations and CIN.
Inhibition of spermatogonial cell proliferation or excessive apoptosis would lead to a decreased sperm count. It has been shown that FD reduces cell proliferation and promotes apoptosis in various cell types (Fenech and Crott, 2002; Guo et al., 2017; Wang et al., 2006; Yuan et al., 2017). Based on previous research, 120 nM folate is the optimal concentration for maintaining genomic stability in vitro in other cell types such as human lymphocytes (Guo et al., 2017; Wang et al., 2006; Wang et al., 2004). In our study, FD significantly delayed the growth of GC-1 cells and enhanced apoptosis. Furthermore, FD promoted an abnormal morphology in GC-1 cells, including an increased size and a flattened and diffuse cell outline. Our results are consistent with several previous studies conducted in other cell types.
However, the proliferation and viability of GC-1 cells cultured with 200 nM folate were markedly affected—despite the lack of morphological changes. This concentration is higher than the physiological folate concentration that had been shown to be sufficient to sustain cell survival and proliferation (Beetstra et al., 2005). Our results demonstrated that GC-1 cells have a high need for folic acid in vitro. One possible cause is GC-1 is an immortalized cell line with rapid regeneration.
Researchers have indicated that male subfertility is associated with poor sperm DNA integrity, including sperm DNA fragmentation, DNA denaturation, and aneuploidy (Ioannou et al., 2019; Panner Selvam et al., 2021). To investigate the effect of FD on the stability of the spermatogonial genome, we used the CBMN-Cyt assay to evaluate cytogenetic damage in GC-1 cells. We also aimed to determine the optimal folate concentration required to reduce the frequency of DNA in spermatogonial cells. The frequency of cells with DNA damage (MN, NPB, or NBud) was inversely and significantly associated with the folate concentration. Each abnormality reached its maximum frequency at the end of the culture period (at day 14) and in folate-free medium.
A plasma folate concentration of 20 nM is not defined as FD in vivo. However, the total genetic damage in the 20 nM folate culture was increased significantly in vitro. Our findings are consistent with a previous study that showed the human plasma physiological folate concentration of 20 nM is not optimal to protect against CIN (Beetstra et al., 2005). It has been reported that these biomarkers, including uracil incorporation into DNA, are usually minimized when cells are maintained in culture medium with a folate concentration >120 nM (Leopardi et al., 2006; Wang et al., 2004). However, in the present study, cells cultured with 200 nM folate still showed a significantly increased frequency of MN, the main biomarker of CIN.
Our results suggest that 200 nM folate is not a safe concentration to protect the genomic stability of GC-1 cells in vitro. The sensitivity to FD may be due to the rapid proliferation of spermatogonia. It must be pointed out, however, that GC-1 cell line is not a standard cell type for the CBMN assay. The CBMN assay in GC-1 cells performed with recommended chemicals should be included in further experiments for optimal folate concentration for maintaining genomic stability in vitro.
In vitro and in vivo evidence has demonstrated that FD is closely related to DNA and chromosome damage, which are thought to be risk factors of pathologies such as cancer, reproductive abnormalities, and congenital defects (Guo et al., 2017; Linhart et al., 2009; Wang et al., 2022; Wang et al., 2004). However, the mechanisms by which FD increases CIN have not been elucidated clearly. Uracil misincorporation is a likely mechanism for FD-induced cytogenetic damage (Blount et al., 1997; Linhart et al., 2009). Simultaneous removal and strand scission of both closely spaced uracil moieties could cause a double-stranded DNA break and lead to chromosome breaks. This eventuality would explain the increased frequency of MN induced by FD. MN can originate from acentric chromosome fragments, acentric chromatid fragments, or whole chromosomes. Only a small proportion of the MN induced by FD is centromere negative and originates from broken chromosomes (Fenech et al., 2011).
It is obvious that the uracil misincorporation model cannot account for all MN. We have provided experimental evidence that FD induces SAC dysfunction through upregulating the expression of core SAC-related genes, which may contribute to mitotic aberrations and chromosome abnormalities.
SAC is a surveillance mechanism that prevents anaphase onset in the absence of kinetochore–microtubule attachment, ensuring equal segregation of chromosomes during mitosis. The key components of this surveillance mechanism, originally identified in budding yeast, include Mad1, Mad2, Mad3/BubR1, Bub1, Bub3, and Mps1 (Benzi et al., 2020; Musacchio, 2015). Among them, Mad2, Mad3/BubR1, Bub3, and CDC20, an essential coactivator of the anaphase-promoting complex/cyclosome (APC/C), assemble the mitotic checkpoint complex (MCC). During prometaphase, SAC is specifically activated at unattached kinetochores of misaligned chromosomes, and the assembled MCC inhibits the APC/C. This avoids anaphase onset and thus prevents chromosome mis-segregation. The expression of various SAC proteins in cells is in equilibrium to maintain SAC function. Researchers have shown that underexpression or overexpression of one or more genes encoding these proteins compromise this equilibrium and lead to SAC dysfunction (Musacchio, 2015).
We found that FD increased the expression of Mad1, Mad2, Bub1, Bub3, BubR1, and CDC20. These results suggest that SAC is disturbed by FD. Guo et al. (2017) recently found that chromosome misalignment was significantly increased by FD in NCM460, a cell line derived from human normal colon mucosal epithelial. Our results support such speculation that FD-induced CMA most likely results from dysregulation of SAC gene expression. Mad2 is the key component of SAC, researchers have found that the amount of Mad2, including Mad1-bound Mad2 and free Mad2, is a crucial determinant of the strength of SAC (Musacchio, 2015).
Mad2 overexpression is deleterious to the ensuing anaphase, leading to chromosome abnormalities such as chromosome mis-segregation, the appearance of broken chromosomes, and anaphase bridges, among other abnormalities (Sotillo et al., 2007). However, overexpression of SAC-related genes does not universally increase SAC activity. Mad1 overexpression by 300–500% reduced the free pool of Mad2 and increased the level of Mad2 at kinetochores; SAC activity was markedly impaired in budding yeast, although the mechanism is unclear (Heinrich et al., 2013).
In addition, CDC20 is an important SAC protein and plays a key role in activating the APC/C. The strength of SAC depends on the degree of CDC20 shielding by the MCC (Heinrich et al., 2013). CDC20 overexpression leads to defective SAC and is associated with premature anaphase promotion, resulting in mitotic abnormalities in various cell lines (Heinrich et al., 2013; Mondal et al., 2007).
In present study, we found the expression of CDC20 was increased significantly in cell-cultured FD medium. Our results are consistent with a previous study demonstrating that CDC20 showed a significant upregulation in liver of mice fed with choline and FD diet (Glen et al., 2015). Our results suggest that there is a distinct possibility that some CIN cells might escape SAC and adapt to grow in FD although CIN induced by FD may be sufficient to induce cell cycle arrest and cell death. These CIN cells may undergo some transformation in their characteristics and overexpress CDC20 to promote their proliferation. Hence, the frequency of BNCs with one or more of the CIN biomarkers (MN, NPB, and NBuds) increased significantly in FD. Our results indicated that the upregulated expression of SAC-related genes resulting from FD promoted GC-1 cells CIN in vitro.
The mechanism underlying FD-induced overexpression of SAC-related genes remains unknown. DNA methylation is an important way to regulate gene expression. It has been suggested that the expression of SAC-related genes is highly sensitive to DNA methylation in the promoter regions (Park et al., 2007). In the present study, we predicted the CpG island location of SAC-related gene promoter regions by using an online tool available at The Li Lab (Laboratory of Molecular Medicine) website. Furthermore, we treated GC-1 cells with different concentrations of 5-AZA, a DNA methylation inhibitor, and found that the expression of Mad2, Bub1, and BubR1 was increased significantly and dose dependently (Fig. 5E). The biological activity of 5-AZA is associated with its incorporation into cellular DNA and/or RNA, with subsequent sequestration of DNA methyl transferases (DNMT) through covalent bond formation between C6 of 5-AZA and cysteine thiolate of DNMTs. Under physiological conditions, this enzyme-DNA/RNA adduct depletes the cells of DNMT activity and causes demethylation of cellular DNA. DNA methylation is one of the best-characterized epigenetic modifications.
DNA methylation in the gene promoter region usually inhibits gene transcription, while demethylation induces gene reactivation and expression. Our results suggest the potential involvement of DNA methylation in the regulation of Mad2, Bub1, and BubR1 expression and upregulation of the expression of Mad2, Bub1, and BubR1 induced by FD may result from demethylation in promoter region of these genes. Further studies are needed to confirm the degree of methylation in specific CpG island of these genes after FD treatment through Bisulfite sequencing (BS-seq). Another potential mechanism of the upregulation of SAC-related gene expression may be associated with the effects of deregulated c-MYC. It has been shown that human BubR1 and MAD2 genes were directly activated by the oncogenic transcription factor c-MYC through E-box sequences in their first introns (Menssen et al., 2007). In present study, we found that FD upregulated the expression of c-MYC in cultured GC-1 cells (Fig. 5D). Further study should be conducted to confirm the correlation between c-MYC and SAC-related genes.
Taken together, our results further demonstrate the important role of folate on spermatogenesis and the proliferation of spermatogonial cells. GC-1 cells were sensitive to FD in vitro because the cells treated with 200 nM folate (higher than physiological folate concentration) showed a significant decrease in viability and proliferation. In addition, FD caused genetic damage to GC-1 cells and increased chromosome abnormalities, which may be the cause of DNA damage and aneuploidy in sperm. Moreover, our results revealed that FD impairs SAC activity by upregulating the expression of several SAC-related genes, which contributes to mitotic aberrations and CIN. These findings establish a novel association between FD and SAC dysfunction. Considering the complexity of the SAC regulatory mechanism and the effects of FD, future investigation is needed to determine the precise mechanism that connects FD and SAC dysfunction. Our findings lay the foundation to further understand the role of folate on spermatogenesis and to formulate reasonable folate supplementation strategies to prevent male subfertility.
Footnotes
Authors' Contributions
Y.L. guided this work and reviewed the article. H.R. and K.W. finished the article. H.R., K.W., and Z.L. helped to finish the experiment. Z.L., X.Z., and M.L. collected relevant information. The final draft was read and approved by all of the writers.
Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by the Natural Science Foundation of the Higher Education Institutions of Anhui Province (China) (KJ2021A0704) and the National Undergraduate Innovative Training Program (202110367050).