
Abstract
Congenital skin disorders are a class of complex genetic diseases that are difficult to diagnose and treat. We developed trio whole-exome sequencing-plus (WES-plus) for detecting de novo mutations and evaluated the use of traditional Chinese medicine (TCM) for treating congenital skin disorders. In this study, we successively performed panel-based next-generation sequencing (NGS) and Trio WES-plus in a child with frequent large blisters. Panel-based NGS revealed no pathogenic mutations. Trio WES-plus for resequencing based on cutaneous keratosis of the palms and feet detected a missense mutation (c.1436T>A,
Introduction
Hereditary dermatoses have a wide variety of similar phenotypes, and related genes are very complex. As patients age and their phenotypes change progressively, it is challenging for clinicians to accurately diagnose and treat such diseases. Epidermolytic palmoplantar keratoderma (EPPK) [OMIM
EPPK is mainly caused by mutations in KRT9 [OMIM# 607606], KRT1 [OMIM# 139350], KRT5 [OMIM# 148040], KRT10 [OMIM# 148080], and KRT16 [OMIM# 148067]. A total of 20 mutations in the KRT1 gene had been identified associated with EPPK. Variations in the keratin genes KRT1 and KRT10 cause epidermolytic hyperkeratosis (Diociaiuti et al., 2014). KRT1 and KRT10 are coexpressed in keratinocytes in the differentiation layer of skin (Wang et al., 2016). KRT1 and KRT10 proteins form the skeleton of keratinocytes (Saeki et al., 2002). The normal expression of these proteins is important for protecting the body from external stimuli, maintaining the normal structure of cells, and enabling adhesion between cells (Ji et al., 2015). KRT1 is mainly expressed in the spinous and granular layer cells of the whole body epidermis (Zeng et al., 2012). Regions of 1A and 2B in KRT1 are the important regions encoding the assembly of keratin filaments in the body. Changes in these regions can affect the formation and stability of keratin filaments, thereby affecting the formation of keratin network structures.
This disease manifests as blisters and erythroderma during infancy. With progressive development of the disease, hyperkeratosis appears and the condition may continue to deteriorate even though the blisters improve and erythroderma decreases. Extensive hyperkeratosis of the skin in adulthood, particularly in curved areas, affects the entire palm surface and can extend to the outside of the fingers and toes. Because different phenotypes are observed at different disease stages, selecting genetic diagnostic methods according to the disease phenotype is difficult and must be addressed to improve the clinical genetic diagnosis of such diseases.
With the development of next-generation sequencing (NGS) technology, panel-based NGS has been increasingly used for diagnosing genetic diseases. However, many types of skin diseases are related to epidermolysis and ichthyosis, which involve hundreds of known and unknown pathogenic genes and similar clinical phenotypes, frequently leading to misdiagnosis. Therefore, many patients with typical characteristics cannot obtain clear genetic diagnostic results. Compared with a gene panel, whole-exome sequencing (WES) technology can capture and sequence the entire exon group with a wider range and greatly increase the probability of detecting disease-causing variations (Hansen et al., 2020). Based on these advantages, WES can identify novel pathogenic genes. Accurate phenotypic information and reliable bioinformatics analysis crucially influence the accuracy of WES and prevent the omission of pathogenic variation. Nonetheless, these two important factors should be further investigated.
In this study, a patient was diagnosed with exfoliative dermatitis, epidermal lax hyperkeratosis, and bullous congenital erythroid ichthyosis. We examined the advantages and disadvantages of different gene detection methods and a new bioinformatics strategy named as trio WES-plus. No pathogenic gene was found using panel-based NGS, whereas a pathogenic mutation in KRT1 of EPPK was screened using trio WES-plus according to the phenotype of palmar keratosis. Choosing appropriate detection and analysis methods when diagnosing genetic diseases is conducive to providing symptomatic treatment and reproductive guidance. The child was effectively treated with integrated traditional Chinese and Western medicine. The parents of the child also successfully gave birth to a healthy second child without this mutation. A novel missense variant of KRT1 was identified and associated with EPPK. This study expands the variant spectrum of KRT1 and sheds light on the importance of prenatal diagnosis and treatment.
Materials and Methods
Study subjects and ethical approval
The Institutional Review Committee of the First Affiliated Hospital of Nanjing Medical University approved this study (2021-NT-70). After explaining the content of the study and possible consequences, we obtained written informed consent from the parents of the proband for various tests to be performed using the proband and parent samples, as well as for prenatal diagnosis. Blood samples were collected from the parents and the proband. Amniocentesis was carried out at 18 weeks of gestation to supply ∼10 mL of Amniotic fluid (AF) for fetal genetic diagnosis. The AF sample was centrifuged at 12,000 rpm for 10 min. After removing the supernatant, the remaining sample was mixed evenly and used for extraction of genomic DNA.
Extraction of genomic DNA
Genomic DNA Extraction Kits (Tiangen Biotech Co., Ltd., China, Beijing, China) were used to extract genomic DNA from the blood of each family member and AF of the fetus. The extracted DNA was dissolved in tris buffer solution. The purity and concentration of DNA were determined using an ND-2000 nucleic acid quantizer (Thermo Fisher Scientific, Waltham, MA) and prepared at a standard concentration of 40 ng/μL DNA template.
Panel-based NGS
The extracted genomic DNA was sequenced as follows. Using interruptase (manufacturer: MGI), genomic DNA is broken up into small fragments of 100–500 bp main band DNA. The interrupted DNA fragments are then screened by magnetic beads, resulting in main fragments that are 150–200 bp in size. The ends are flattened, and the base “A” is added at the 3′ end to make the DNA fragments ligated to special junctions with “T” bases. After the hybridization reaction, the DNA was washed and eluted using the probe for the reaction, followed by the ligation-mediated PCR of the Captured samples. Agilent 2100 Bioanalyzer and BMG examined the libraries for fragment size and concentration, and libraries requiring different data amounts were pooled and quantified after passing the test; then the pooled libraries were subjected to single-stranded cyclization. Using a high-throughput sequencer called the BGISEQ-500, the libraries were sequenced for 50 cycles after being created using DNB, and the raw sequencing results were then read out.
Detection genes include KRT5 [OMIM# 148040], KRT14 [OMIM# 148066], ITGB4 [OMIM# 147557], ITGA6 [OMIM# 147556], PLEC [OMIM# 601282], LAMB3 [OMIM# 150310], COL17A1 [OMIM# 113811], LAMC2 [OMIM# 150292], LAMA3 [OMIM# 600805], COL7A1 [OMIM# 120120], ALOX12B [OMIM# 603741], and ALOXE3 [OMIM# 607206].
Target-sequence capture and sequencing
WES was performed on samples from all participants in this study (proband and parents). Genomic DNA (1 μg) was sheared into fragments of ∼170 bp, and fragments containing coding exons, flanking intronic regions, and promoter regions were captured using an Agilent SureSelect Target Enrichment Kit (Agilent Technologies, Santa Clara, CA). After generating DNA nanoballs from the ssDNA circle using rolling circle amplification, the enriched libraries were sequenced on an MGISEQ-500 platform (BGI, Inc., Shenzhen, China) according to the manufacturer's protocols.
Sequence analysis
After sequencing, the data were downloaded and analyzed. First, the sequencing quality of the raw data (raw reads) was evaluated to remove low-quality and contaminated reads. Second, the sequencing reads were aligned to the human reference genome hg19 using Burrows–Wheeler Aligner with default parameters, and the capture effect of the sequence was evaluated. Genome Analysis Toolkit software was used to query single nucleotide variant (SNV) and insertions and deletions to detect base polymorphisms in the target region. All identified variants were annotated using the following four databases: 1000 Genomes Project (
Bioinformatics analysis of KRT1 gene
According to the disease phenotype, phenotypic terms were searched in the Human Phenotype Ontology (HPO) database (
The disease phenotype caused by the gene was compared with the child's phenotype to detect overlap. Genes that were highly matched to the child's clinical phenotype were retained. SIFT, MutationTaster (
Sanger sequencing to verify gene variations
Primers were designed online using Primer Design software Primer BLAST from NCBI. The reaction system of PCR (50 μL system) contained 1 × buffer, 1.5 mM MgCl2, 5 U Taq DNA polymerase, 2 mM dNTP mixture, 1.2 μM forward primer (5′-AAG CTG AAT GAC CTG GAG GAT G-3′), and 1.2 μM reverse primers (5′-TCA GTT TCG GGG CAC ATT C-3′). The thermocycling conditions (65–60°C touch down) were as follows: initial denaturation at 94°C for 10 min, followed by 12 cycles at 94°C, 60°C, and 72°C for 40, 30, and 60 s, respectively, and then 28 cycles at 94°C, 55°C, and 72°C for 40, 30, and 40 s, respectively; final extension was performed at 72°C for 10 min. After amplifying the genomic DNA of the child and his parents, the products were subjected to Sanger sequencing. The sequencing results were compared with the reference sequence of KRT1 (NM_006121.4).
Combination therapy and prenatal diagnosis
Long-term oral administration of acitretin A and topical emollients, retinoic acid, keratinizing agents, and vitamin D analogs has been conducted to treat this disease. In addition, the patient was administered with traditional Chinese medicine (TCM). In the last several years, the child has consumed boiled green tea, cattail, and jasmine to improve the clinical symptoms.
During the second pregnancy, amniocentesis was performed at 18 weeks of gestation. Karyotype analysis of the AF cells was performed as the gold standard method. Short tandem repeat loci were evaluated to detect maternal blood contamination of the AF DNA, and Sanger sequencing was performed to determine whether the fetus carried the mutation in KRT1, c.1436T>A (p. Ile479Asn). Skin development was monitored after birth.
Results
Clinical description of patient
The skin on his entire body of our young male patient has been covered with moist cotton wadding since birth, as large blisters frequently erupt across the body. Thick white skin covers his scalp, hands, and soles of the feet. The patient was initially diagnosed with bullous epidermolysis. At the age of 4 years, the skin darkened and progressively fell off, with blisters occurring in the nonblackened areas, particularly on the skin of the palms and soles of the feet, which were still undergoing keratinization. At 12 years of age, thick skin was observed on the palms, soles, and knees. The foot bends and elbow joints were circularly keratinized (Fig. 1). Although the number of blisters decreased as the child aged, a rough layer of skin remained on the scalp. There was an aftereffect of sweating, and the sweat emits a strong odor.
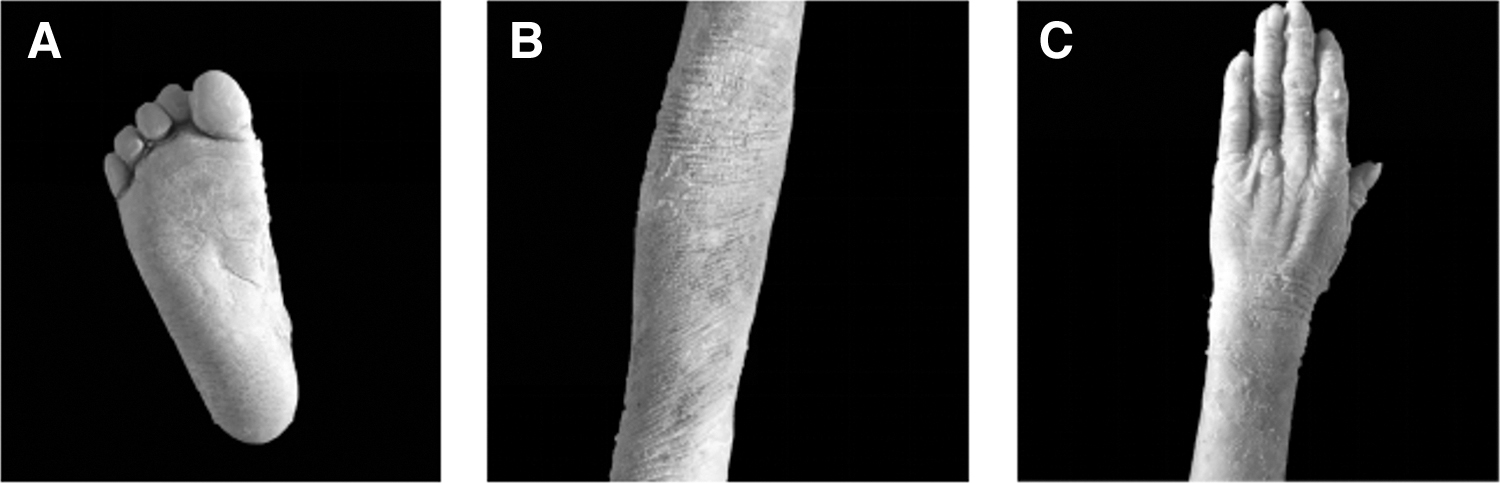
Photographs of the proband's clinical phenotype.
Panel-based NGS in epidermolysis bullosa
The coding region and neighboring ±10-bp intron region were captured in the gene panel, and 31 variable sites were obtained. These sites were classified as benign or likely benign based on population frequency screening and ACMG guidelines (Supplementary Table S1). No pathogenic or likely pathogenic mutations were identified within the detection range of the panel.
Potential pathogenic variation detected using trio WES-plus sequencing combined with Sanger sequencing and verification of pathogenicity based on ACMG guidelines
The entire exon group of the child and his parents were captured and sequenced using an NGS platform. Approximately 22,000 nucleotide variations were found, and the KRT1 heterozygous site (c.1436T>A, p. Ile479Asn) was screened. The Sanger sequencing results were consistent with those of NGS. The child showed base T/A heterozygosity at nucleotide site 1436, whereas both parents had the normal base T (Fig. 2). Mutations in KRT1 cause EPPK (Terron-Kwiatkowski et al., 2004), which is consistent with the child's phenotype.
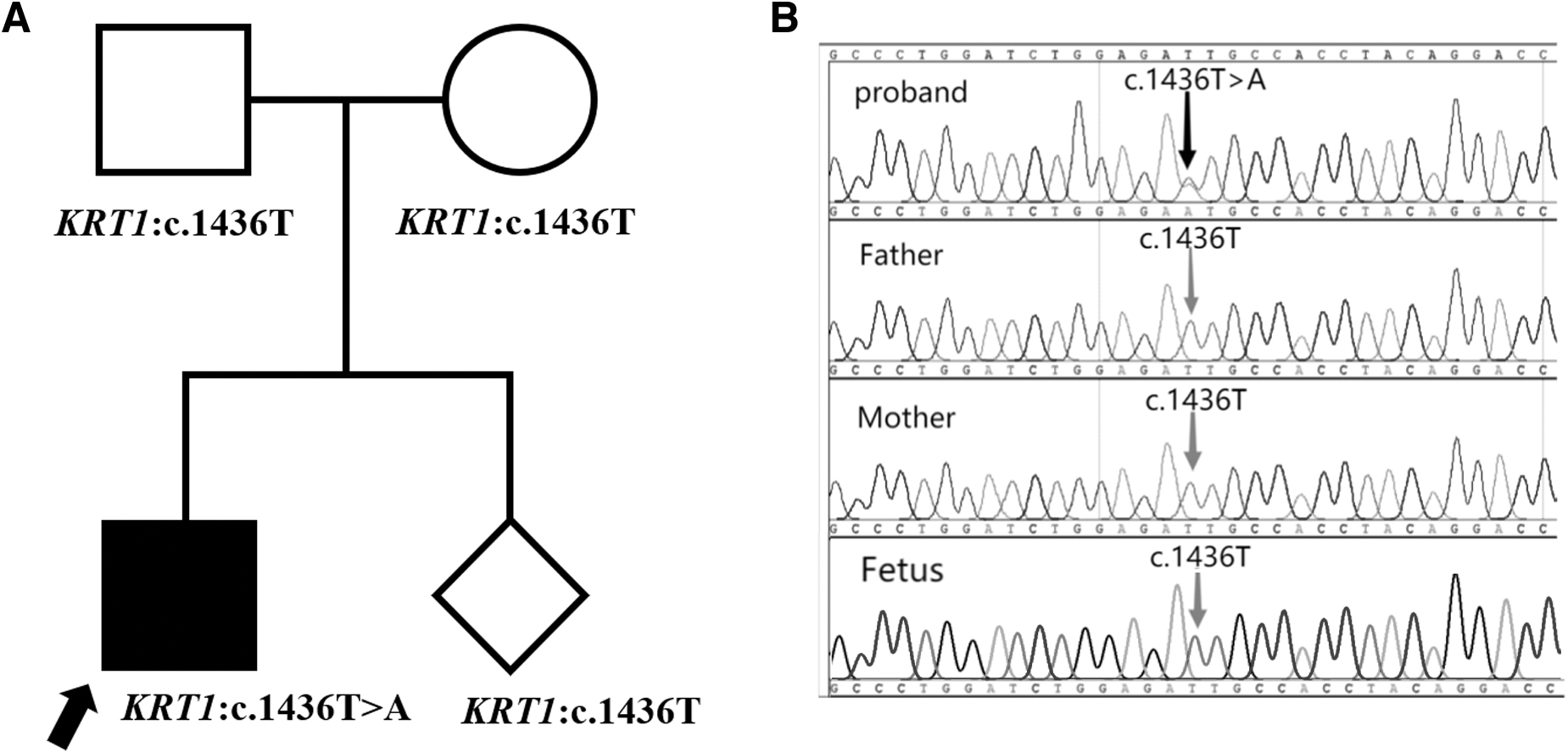
Possible pathogenic variation.
According to ACMG Standard Guidelines, KRT1 was an autosomal dominant gene de novo in this family (PS2). The detected variant has not been reported in the gnomAD_exome, ExAC, or 1000 genome databases and it is not polymorphic (PM2_Supporting). The function of the protein encoded by the gene containing the variant was predicted using various bioinformatics software programs (SIFT, MutationTaster, Polyphen2, etc.). The results indicated that the variant was harmful. Prediction of nucleic acid conservation (GERP++, phyloP, etc.) (Table 1) indicated that the variant was conserved (PP3). The new missense mutation (c. 1436T>A) was found at the 479th amino acid residue from isoleucine to asparagine. Different changes in the 479th amino acid residue from isoleucine to threonine and phenylalanine were confirmed as pathogenic (PM5). The child's phenotype was highly consistent with autosomal dominant EPPK (PP4). According to existing evidence (PS2+PM5+PM2_Supporting+PP3+PP4), the mutation is likely pathogenic (Fig. 3).

Evaluation of the pathogenicity of a mutation (c.1436T>A) in KRT1 according to ACMG guidelines. ACMG, American College of Medical Genetics.
The Pathogenicity of the KRT1 Variant
Analysis of the NM_006121.4 transcript as a reference showed that the KRT1 gene contains 9 exons, with c.1436T>A in exon 7. The 479-Ile site is highly conserved in various species, such as humans, rats, mice, dog, and so on. (Fig. 4). SAAPdap (Single Amino Acid Polymorphism Data Analysis Pipeline) and SAAPpred (Single Amino Acid Polymorphism Predictor) were used as structure models to evaluate the effect of Ile479 variant. As shown in Figure 5, the de novo mutation (c.1436T>A,

Conservation of Ile479 in various mammalian homologs, including humans, rats, mice, dogs, horses, rhesus monkeys, and cattle. The box represents the mutated residue.

Before and after mutations of KRT1 and KRT10 binding site.
Clinical validation and guidance on childbearing based on prenatal diagnosis
Considering that EPPK is an autosomal dominant genetic disease, it is thought to occur de novo; however, the possibility of parental gonadal mosaicism cannot be ruled out. Therefore, for the next pregnancy in the family, prenatal diagnosis was recommended to exclude the same cause of disease. Genetic diagnosis of AF samples during pregnancy was performed using Sanger sequencing. The c.1436T>A (p. Ile479Asn) mutation KRT1 was not detected in the fetus. Thus, the parents chose to continue the pregnancy and eventually gave birth to a healthy girl. The girl's phenotypic profile was normal according to a recent follow-up, which is consistent with the results of the prenatal diagnosis (Fig. 2).
Combination therapy effect of TCM and western medicine
Clear genetic diagnosis of EPPK can guide medication use and improve the life of the child. However, there is no specific treatment for this condition. We treated the affected child with a combination of TCM and Western medicine. In the last several years, the consumption of boiled green tea, cattail, and jasmine alleviated the dryness of the child's skin and reduced dandruff and body odor.
Discussion
Mutations in KRT1, which is located on chromosome 12q11–q13 (Smith, 2003), mainly cause abnormalities in the structure, arrangement, or aggregation of keratin intermediate filaments (KIFs), leading to excessive keratinization accompanied by intracellular edema and resulting in clinical manifestations of EPPR. KIFs are an important part of epidermal cells. They polymerize to form cytoskeleton networks around the nucleus and can form Tonofibril bundles that pass through the cytoplasm to establish desmosomes or semi-desmosomes between cells, playing a vital role in the stability of cell structure and adhesion between cells.
The keratins are divided into two sizes based on molecular weight: the low molecular weight acidic type I (KRT9-KRT20) and the high molecular weight basic type II (KRT1-KRT8) (Moll et al., 1982). Both of acidic and basic keratins consist of a central α-helical coiled-coil rod domain of 310 to 315 amino acids in size. The central alpha-helical rod domain is composed of four helical segments (1A, 1B, 2A, and 2B) that are interrupted by three short nonhelical flexible linker regions (L1, L12, and L2) (Fig. 6). The rod domain is composed of repeats of seven amino acid residues (a-b-c-d-e-f-g)n, in which positions “a” and “d” are occupied by hydrophobic residues that are considered crucial for the coiled-coil formation. The start of the 1A rod domain and the end of the 2B rod domain, the so-termed helix initiation and helix termination peptides (HTP), respectively, comprise 20 amino acid sequence motifs that are most highly conserved among the different keratins. These motifs play a critical role in the overlapping interactions during KIF assembly (Chamcheu et al., 2011).
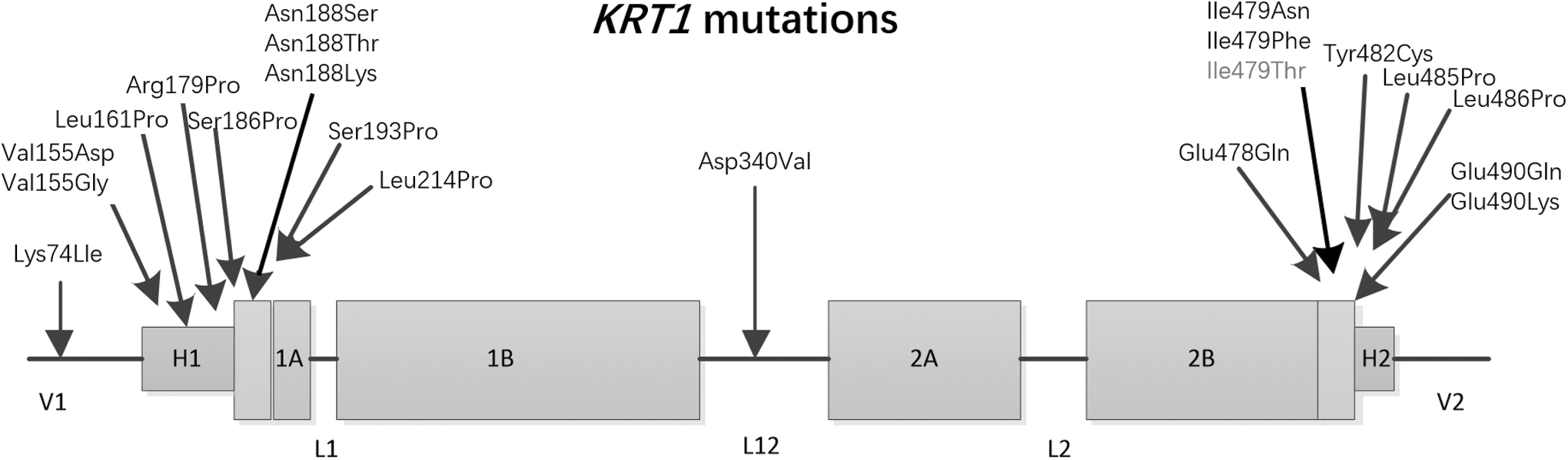
KRT1 protein structure showing reported mutation sites.
There are 20 reported pathogenic variants of KRT1, 18 of which are in the intermediate filtration domain (180–493) (Hesse et al., 2001; Ji et al., 2015; Zaki et al., 2018). Mutations in these regions result in the most severe form of epidermal fragility syndrome, indicating that this domain has a critical and conserved function. The 2B rod domain (HTP) of KRT1, from the 478th amino acid to the 490th amino acid, is currently considered a highly conserved region of intermediate silk proteins (Sung et al., 2013). The mutation site (c.1436T>A,
A critical aspect influencing the detection rate of disease is the selection of an effective genetic testing technique (Gao et al., 2016; Meienberg et al., 2016; Sun et al., 2015), which is also closely associated with the accuracy of the clinical diagnosis of phenotypes. Our patient was diagnosed with exfoliative dermatitis, EPPK, and bullous congenital erythroid ichthyosis present since birth. The phenotype of palmoplantar keratosis which is a typical phenotype of EPPK caused by the KRT1 mutation had not been observed. As KRT1 was not included in the gene panel used to diagnose the child, no positive findings were obtained using this method. Then, we performed WES on three people (the affected child and his parents). WES is appropriate for analyzing difficult cases with complex clinical manifestations and an ambiguous disease diagnosis (Hansen et al., 2020), as well as for those showing negative results in panel-based NGS, such as the child in this case (Ewans et al., 2018; Sun et al., 2015). A de novo mutation in KRT1 of the child was detected using WES.
Bioinformatic analysis has crucial effects on the detection rate of disease. Two methods of data analysis were used in this case. In the first method, regardless of the child's phenotype, the analysis focused on protein-truncating or stop-loss variants in genes. This method consists of five steps. The first step is eliminating variations with frequencies greater than 0.005 in the population. Second, variants that were computationally predicted to be benign are removed, whereas pathogenic variants are retained. The third step is to prioritize severe variations such as truncation, nonsense, and frame-shifting variants. The fourth step is analyzing the pathogenicity of variations according to ACMG guidelines. The fifth step is checking the OMIM, ClinGen, GeneReviews, and other databases to determine whether the phenotype and inheritance match for the remaining variations. Unfortunately, no positive results were found for the first method.
Therefore, we modified our analysis strategy to focus on screening for variations related to the child's phenotype. The first step of the second method is to determine the potential genetic model based on the child's medical history and family history and then filter out SNVs that fit the genetic model from the data. The second step is to search for standard phenotypic terms in HPO according to all phenotypes of the child, find all genes related to the phenotypes in OMIM and GeneCards, and match the above-reserved genes. The third step is to retain the mutation sites with population frequencies less than 0.005. The fourth step is ranking the variations according to ACMG guidelines. To ensure that the final retained variation matches the patient's phenotype, we re-queried OMIM and other databases. This analysis strategy emphasizes the importance of combining clinical and genetic data to make useful prognostic predictions.
Using the second method, we screened 4880 SNVs from 21,221 SNVs detected using trio WES plus based only on the genetic mode; 23% of the results were retained, greatly improving the efficiency of data analysis. Based on the phenotype, we searched for standard phenotypic terms in HPO, such as “HP:0000982 palmoplantar keratoderma,” “HP: 0007543 Epidermal Hyperkeratosis,” and “HP: 0007475 Congenital Bullous Ichthyosiform Erythema,” and used these terms to search and summarize relevant genes in GeneCards and OMIM. After matching with the 4880 SNVs, 2614 SNVs were screened, and 12.3% of the results were retained. We identified 660 SNVs (3.1%) with a population frequency less than 0.005 according to analysis of the mutation frequency using the gnomAD, 1000 genomes, ExAC, dbSNV, and other databases. The 660 candidate SNVs were graded according to ACMG guidelines and checked on the OMIM website to evaluate their degree of match with the child's phenotype. Finally, a de novo missense mutation (c.1436T>A, p. Ile479Asn) was identified in the coding region of KRT1 (Fig. 7).
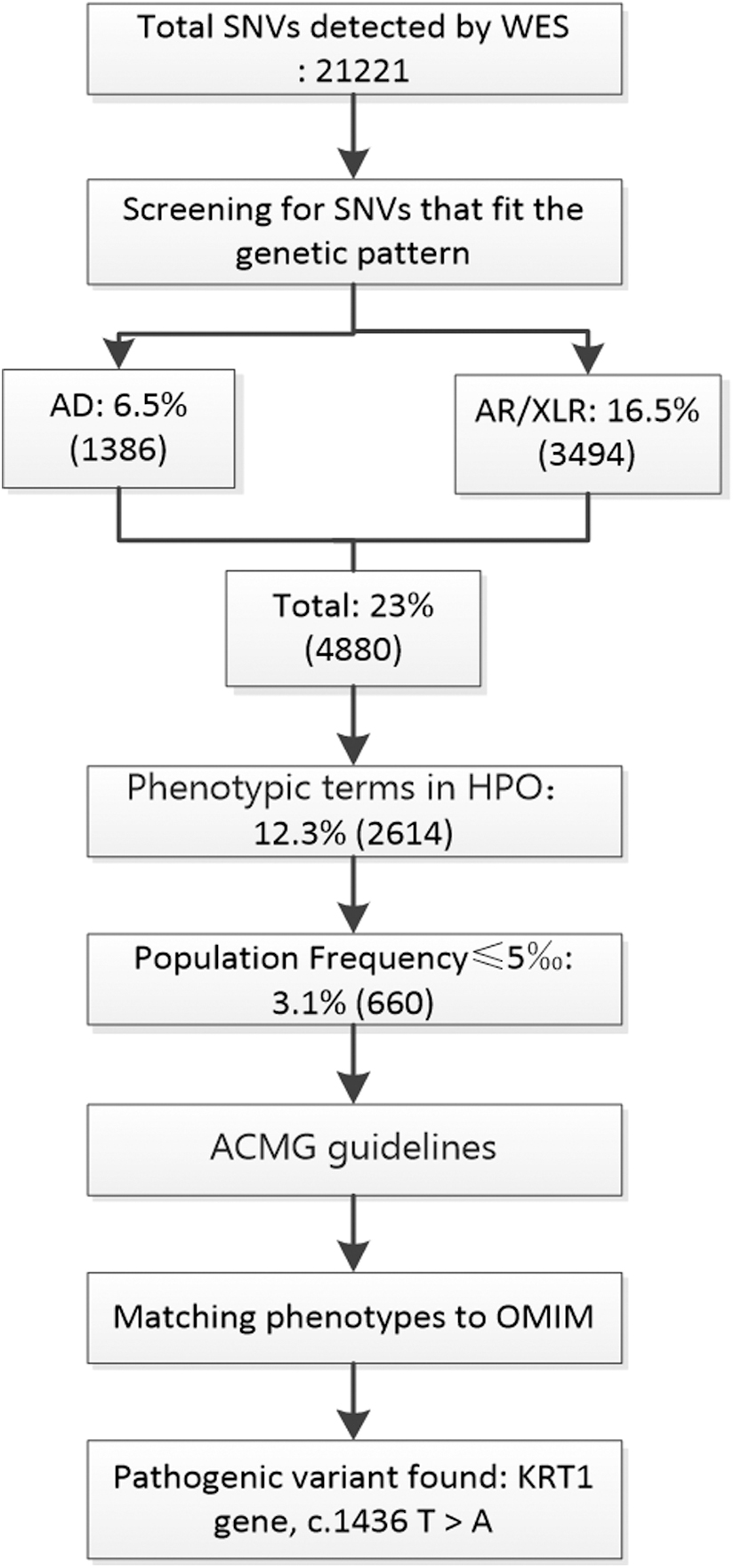
WES data analysis. WES, whole-exome sequencing.
Identification of the pathogenic variation in the child provided a basis for genetic counseling and treatment. The child was administered combination therapy of TCM and Western medicine. Green tea can be consumed daily with almost no side effects, and tea polyphenols can improve skin symptoms. Tea polyphenols, derived from pure natural green herbs, is inexpensive and easy to obtain. Pathological damage to the skin was significantly improved by drinking green tea daily. Jiujie calamus can alleviate muscle redness and inflammation caused by skin injury. Jasmine contains numerous aromatic hydrocarbons, such as jasmine and linalool, as well as volatile oils that can moisturize the skin and improve body odor. TCM therapy has substantially improved the lives of people with skin problems.
Trio WES plus technology can provide a precise diagnosis and predict a patient's prognosis. Guidelines should be provided to families before giving birth. Amniocentesis was performed after the proband's mother was 17 weeks pregnant to determine whether the fetus had the same mutation. Amniocentesis combined with Sanger sequencing is an optimal method for detecting point mutations. According to a recent follow-up, the second child was a healthy girl.
Conclusions
We identified a novel heterozygous missense mutation in KRT1 (c.1436T>A, p. Ile479Asn), which is a likely pathogenic variant causing EPPK. After a clear diagnosis, the patient received effective combination therapy of TCM and Western medicine to improve the symptoms, and the family obtained a healthy child through prenatal diagnosis. Our results expand the genotypic spectrum of EPPK and enrich the HGMD mutation database, as well as provide a strategy for using trio WES plus to diagnose and treat patients showing negative results in other genetic diagnosis technologies.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the third session of the Key Provincial Talents of Maternal and Child Health Program in Jiangsu Province and “333 High-Level Talents Training Project” (2022-3-25-009).
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.