
Abstract
Developmental dysplasia of the hip (DDH), characterized by acetabular deformity that manifests from loose ligaments to complete dislocation of the hip, can cause notable pain and dysfunction and lead to hip dislocation, secondary fractures, scoliosis, and osteoarthritis of hip. Variants in FLNA may produce a spectrum of malformations in multiple organs, especially the skeleton. This study aimed to identify the genetic etiologies of DDH patients and provide genetic testing information for further diagnosis and treatment of DDH. We recruited a Chinese woman with DDH and her family members. Whole-exome sequencing was used to identify the patient's genetic etiologies. Protein models were used to analyze the pathogenic mechanism of the identified variants. A novel variant (c.3493T>G, p.C1165G) of FLNA was detected. The structural models of the mutant FLNA protein indicated that the variant would lose its sulfhydryl side chain and destroy the attraction between benzene rings and sulfhydryl. We reported a novel variant (c.3493T>G, p.C1165G) of FLNA in a Chinese woman with DDH. Our research outcome enriches the gene pool for hip dysplasia and emphasizes the pathogenicity of sulfhydryl side chain disruption in FLNA.
Introduction
F
Previous studies have already established the association of FLNA variants with certain conditions, such as otopalatodigital syndrome (Robertson et al., 2003), Melnick-Needles syndrome (MNS) (Luo et al., 2023; Robertson et al., 2003), Terminal osseous dysplasia, and developmental dysplasia of the hip (DDH) (Sun et al., 2010).
DDH, characterized by a range of deformities caused by acetabular dysplasia, is a prevalent condition affecting the hip joint, particularly in women, with an incidence of ∼17 cases per 1000 live births in China (Xu et al., 2022). Pathologically, during the perinatal period, the femoral head loses its normal relationship with the acetabulum within the joint capsule (Harsanyi et al., 2020b), impeding proper development and leading to ligament laxity and subsequent hip joint dislocation. This condition often causes substantial pain and functional impairment for the affected individuals (Schmitz et al., 2020). As the patient grows and the disease progresses, there is a likelihood of complete dislocation of the hip joint.
Consequently, compromised muscle development and impaired blood supply occur on the affected side compared to the unaffected side. This further hampers the development of the femur on the affected side, resulting in a significantly shorter length of the affected lower limb relative to the healthy side. The subsequent long-term imbalance in mechanical stress renders the patient susceptible to femoral neck fractures and osteoarthritis of the hip joint (Bankaoğlu, 2019). In severe cases, involvement of the spine may lead to the development of scoliosis and kyphosis. Notably, abnormal changes in the hip joint can often be detected in neonates, indicating the early onset of DDH in adult patients.
Previous study has reported associations between DDH and 16 genes (Harsanyi et al., 2020a), including ATP2B4 (Basit et al., 2017), HSPG2 (Basit et al., 2017), and BMP2K (Zhao et al., 2017). However, the FLNA gene has not been reported in this review, indicating a gap in the gene pool for this disease. Furthermore, despite extensive structural and functional studies on the FLNA gene (Zhou et al., 2021), the specific mechanisms by which its variations contribute to bone developmental disorders remain unclear. In this study, we discovered a new variant (c.3493T>G, p.C1165G) of the FLNA gene that is associated with the phenotype of DDH in a Chinese woman. This finding adds to our understanding of the genetic complexity of DDH as a heterogeneous disease. Furthermore, we investigated the protein structural changes to shed light on the underlying pathogenic mechanism.
Materials and Methods
Subjects
The study protocol was approved by the Ethics Review Committee of Xiangya Hospital of Central South University in China (approval no.: 202103427), and all participants signed informed consent. The subjects of this study were a Chinese family with DDH. The proband (II:2) presented to Orthopedics Department of Xiangya Hospital in 2021, as well as her family members, parents (I:1 and I:2), husband (II:1), and son (III:1) (Fig. 1A). The family medical history, ancillary examinations, and blood samples of the subjects were collected for analysis.

Pedigree, symptom, and Sanger sequencing of probands.
DNA extraction
Genomic DNA was extracted from the patients' peripheral blood lymphocytes using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA).
Whole-exome sequencing and Sanger sequencing
The skeletal dysplasia-related genes are summarized in Supplementary Table S1, and we adopted the whole-exome sequencing (WES) technology to screen these variants listed in the table. The specific procedure is illustrated in Figure 2. The main part of WES was provided by the Berry Genomics Company Limited (Chengdu, China), as previously described (Jin et al., 2022). All the exomes were captured by Agilent Sure Select Human All Exon V6 kits and sequenced by Illumina HiSeq X Ten platform.
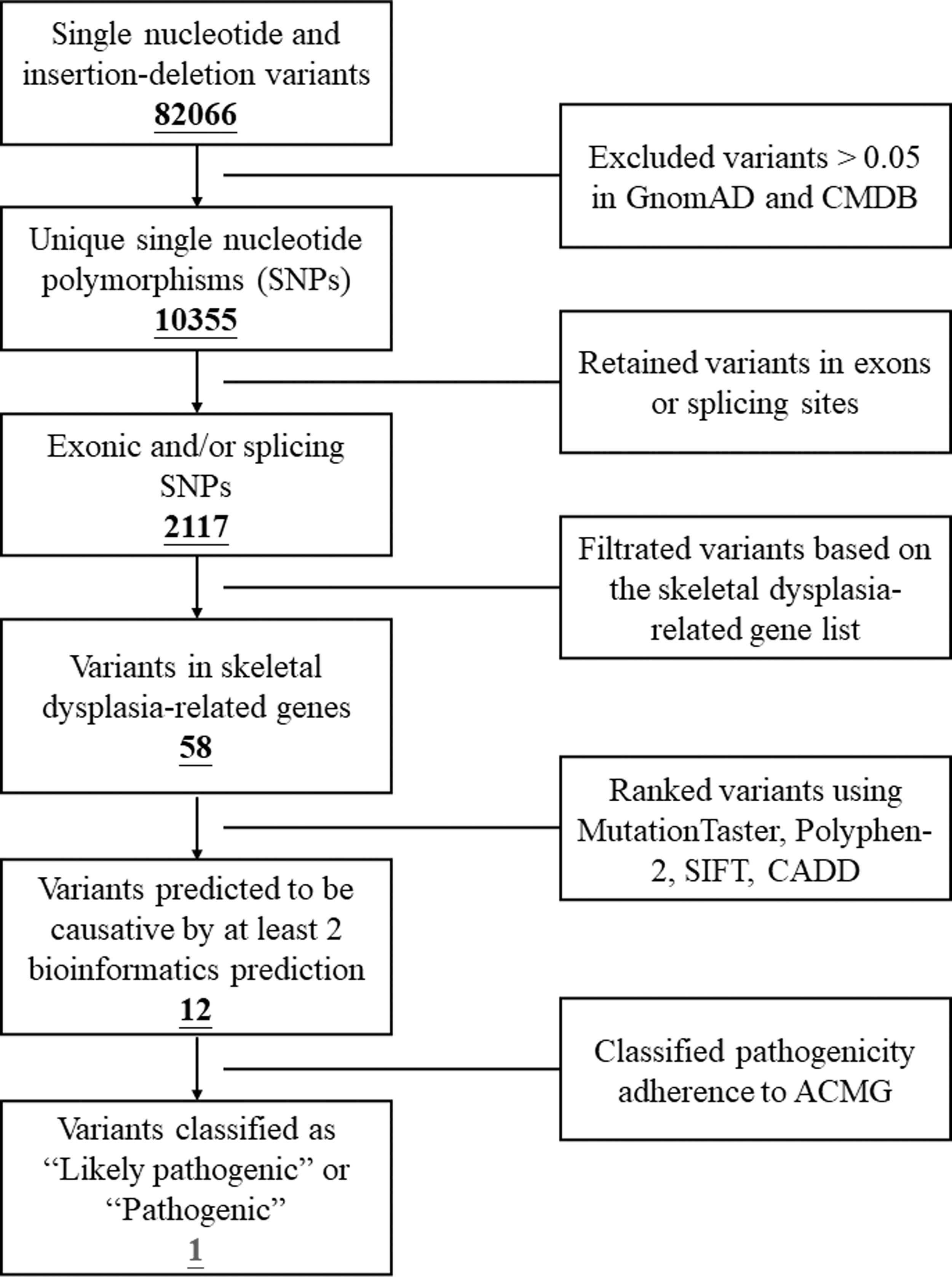
Workflow diagram depicting the processes of WES and Sanger sequencing. WES, whole-exome sequencing.
The programs of Polyphen-2 (
Primer pairs were designed by software Primer Premier 5.0 (Qin et al., 2009) (FLNA f: 5′-AACATCCTCTTCGCTGACAC-3′; FLNA r: 5′-ACCGTGGTCCTGGATGTA-3′). The sequences of primers could be provided on request.
Mutant protein modeling
The filamin A protein structure of human (AF-P21333-F1) was downloaded from UniProt Protein Database (
Results
Case description
The proband in this case study was a 35-year-old woman who was admitted to our hospital due to hip dysplasia (Fig. 1A). She had experienced difficulties with standing since the age of 1, but no specific treatment was administered at that time. At the age of 34, she started to experience pain in her left hip, which was accompanied by significant pain during movement. X-ray imaging revealed several abnormalities: the left acetabulum showed reduced development compared to the opposite side, the femoral head was flattened and displaced upwards, the joint space was narrowed, and subchondral bone sclerosis was observed in both the acetabulum and femoral head. In addition, the epiphysis of the femoral head appeared smaller and displaced outward and upward. The affected side of the femur and pelvis exhibited underdevelopment, resulting in a tilted pelvis toward the healthy side (Fig. 1B). Furthermore, scoliosis was observed (Fig. 1C).
Physical examinations indicated gait claudication, tenderness, limited movement in the left hip joint, positive Patrick's sign, and longitudinal percussion pain. The left lower limb was found to be 8 cm shorter, and the range of motion in the left hip joint, including adduction and abduction, was significantly restricted. Consequently, the patient was diagnosed with left congenital hip dysplasia, left congenital hip dislocation, and scoliosis. She underwent simultaneous hip arthroplasty and exhibited favorable postoperative recovery. No other family members were affected by these conditions.
Genetic analysis
WES yielded 10.12 Gb data with 99.8% coverage of the target region, and 98.9% of the target was covered over 10 × (Supplementary Data supplementaldata). We then performed a genetic analysis on skeletal dysplasia-related genes (Supplementary Table S1) to filter the 228 remaining variants of the patient, and a set of 5 variants in 5 genes was identified. Eventually, a novel variant (c.3493T>G, p.C1165G) of FLNA with DDH was discovered (Table 1). Sanger sequencing was used to verify the FLNA variant in proband and detect the genotype of other family members (Fig. 1D). The FLNA variant identified in proband was de novo and was not detected in other family members.
Variants Identified in the Patient by Whole-Exome Sequencing in Combination with the Filtration of Genes in Skeletal Dysplasia-Related Genes
B: benign; D: disease causing.
CMDB, Chinese Millionome Database; MNS, Melnick-Needles syndrome; XLD, X-linked dominant; XLR, X-linked recessive.
Amino acid sequence alignment analysis indicated the comparatively conservative in evolution of the FLNA variant site, p.C1165 (Fig. 1E). Based on the human filamin A protein modeling, p.C1165 residue can interact with the benzene rings of the p.F1181 and p.F1243, while the variant p.C1165G damaged these ionic bonds (Fig. 3). Thus, we reasoned that the FLNA variant (c.3493T>G, p.C1165G) was the genetic pathogenic factor of the proband.

Comparison of protein structures at the mutation site between the wild type and mutant type
Discussion
In this study, we used WES and Sanger sequencing to examine the presence of possible genetic damage in a patient with DDH, and discovered a new FLNA variant (c.3493T>G, p.C1165G). The novel variant of FLNA (c.3493T>G, p.C1165G) was not identified in GnomAD and CMDB.
In accordance with the ACMG guidelines, we classified the pathogenicity of the FLNA variant as “likely pathogenic” for one strong evidence (PS2), one moderate evidence (PM2), and one supporting (PP3) evidence (Table 1): (1) De novo variant (biological parental samples testing negative) in a patient with the DDH and no family history (PS2; Fig. 1A); (2) absent from controls in GnomAD and CMDB (PM2); (3) predicted for a deleterious effect by more than half of mainstream databases: MutationTaster, SIFT, and CADD predicted that the variant was disease causing (a variation score more than 15 is considered to be pathogenic in CADD; PP3).
Figure 1D shows that thymine was altered to guanine at position 3493 of the coding sequence of the FLNA gene; as a consequence, cysteine was substituted by glycine at position 1165 of the amino acid. Before the variant, a π-π stacking was formed between the benzene ring at F1243 and the benzene ring at F1181 of FLNA. Meanwhile, the π bond of the benzene rings is negative central, and sulfur is electronegative. The attractive interactions arise from the polarity of two groups, respectively. A triangular structure was constructed by those three interactions. In comparison, after the variant, the sulfhydryl side chain in C1165 is destroyed, resulting in the disappearances of original two pairs of attraction (Fig. 3). As a result, the space structure and stability of filamin A are likely to be impaired as well.
The FLNA protein is a 280 kDa subunit homodimer composed of an actin binding domain and 24 immunoglobulin (Ig)-like repeats (R). It has been reported to play a role of inhibiting osteogenic differentiation in vitro (Yang et al., 2022) and promoting osteoclast differentiation (Hu et al., 2017; Leung et al., 2010). In this study, the structural domain where the variant is located is FLNA-Ig10. The missense variants at Ig-10 have often been reported to be associated with early bone dysplasia such as ear-finger syndrome spectrum diseases, frontal metaphyseal dysplasia (FMD), and MNS.
Meanwhile, the stability of this domain mainly depends on the disulfide bonded lattice and cysteine adducts (Page et al., 2011). Following FLNA variant in our patient, it was found that the two pairs of attraction at C1165 disappeared, which might reduce the local stability of Ig-10, thereby altering the regulation of FLNA protein on osteogenesis and osteoclasis and resulting in bone dysplasia.
The known variants near our identified variant and their phenotypes are listed in Table 2. Most of them are associated with the diseases involving bone development. Among them, MNS is an extremely rare osteochondral dysplasia, which mainly occurs in long bones, vertebrae, and ribs; it has also been associated with respiratory system diseases, speech disorders, and chewing problems (Jung et al., 2012). The patients typically have short stature, accompanied by scoliosis, partial dislocation of the joints or bone defects. FMD is a rare genetic disease presenting with morphological abnormalities in bone and extraskeletal tissues (Weiss et al., 1976), which can be characterized by prominent frontotemporal bone dysplasia, rib dysplasia, joint contracture, and systemic muscular dysplasia (Weiss et al., 1976). In this study, our patient was found to have bone dysplasia and dislocation of the hip joint, scoliosis, as well as kyphosis.
Variants and Phenotypes Reported Near Our Mutation
Similar to the diseases mentioned above, the patient also had the problem of bone and joint dysplasia. Scoliosis is a three-dimensional deformity of the spine, involving sequence abnormalities in the coronal, sagittal, and axial positions. Patients with congenital scoliosis usually have an onset age of <10 years (Cunin, 2015) and are associated with other congenital genetic diseases such as DDH. Previous case report has described phenotypes involving variants in the FLNA gene, accompanied by DDH and congenital pes torus (Calcaterra et al., 2018), suggesting that DDH can coexist with other skeletal developmental abnormalities. In our study, the patient exhibited both scoliosis and DDH, suggesting that the scoliosis may be attributed to the genetic variant.
Furthermore, during the process of growth and development, the imbalanced stresses caused by DDH can lead to compensatory spinal deformities, resulting in scoliosis. Ultimately, the phenotype of this patient may be the result of a combination of congenital genetic factors and the impact of imbalanced stresses during growth and development.
Conclusion
This study reported a DDH case and identified a novel FLNA variant (c.3493T>G, p.C1165G) by WES and Sanger sequencing. Bioinformatics analysis of phenotypic and functional changes in the patient suggests that FLNA variants are pathogenic and associated with DDH. Our research results enriched the spectrum of FLNA variants, revealed the pathogenicity of sulfhydryl side chain breakage in FLNA, and further strengthened the correlation between the phenotype and genotype of DDH, thereby providing a strong basis for exploring the consequences of DDH-related variants in the future.
Footnotes
Acknowledgments
We thank all subjects and medical personnel in this study.
Authors' Contributions
Conceptualization (J-.Y.J.); Methodology (Y-.L.L. and Q.W.); Funding acquisition (J-.Y.J. and J-.Y.T.); Data curation (J-.Y.J.); Investigation (Y-.L.L., Q.W., M.W., S-.H.C., J-.Q.H., R.X.); Resources and Software (M.W., S-.H.C., J-.Q.H., and R.X.); Supervision (J-.Y.J. and J-.Y.T.); Visualization (Y-.L.L. and Q.W.); Writing-original draft (Y-.L.L. and Q.W.); and Writing-review and editing (J-.Y.J. and J-.Y.T.). All authors have read and agreed to the published version of the article.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China (82272508, 82102527, and 82200776), Key Research and Development Program of Hunan Province (2022sk2034), the Natural Science Foundation of Hunan Province (2022JJ30058, 2020JJ5785, and 2021JJ40968), the Provincial Science and Technology Department Foundation of Hunan (2021ZK4218).
Supplementary Material
Supplementary Table S1
Supplementary Data supplementaldata
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.