
Abstract
Liver fibrosis (LF) is a clinical disorder characterized by inflammation and excessive accumulation of extracellular matrix (ECM). This study investigates the effects of the antifibrotic compound pirfenidone (PFD) on improving LF through histological changes and modulation of eukaryotic translation initiation factor 6 (eIF6), P311, and transforming growth factor beta (TGF-β) in rats with bile duct ligation (BDL)-induced LF. Rats received daily doses of PFD (200 and 500 mg/kg) for 4 weeks. The study encompassed biochemical, pathological, and immunohistochemical (IHC) analyses. mRNA levels of eIF6, P311, TGF-β, ECM deposition, hepatic stellate cell (HSC) activation, and inflammatory mediator genes were measured by RT-qPCR. Protein levels of eIF6, P311, and TGF-β were detected by western blotting. Compared with the BDL group, PFD dose-dependently reduced hydroxyproline content, liver index, biochemical parameters, fibrosis score, and fibrosis area. PFD also modulated BDL-induced hepatic inflammation, ECM accumulation, and HSC activation. IHC staining of Ki-67 and hepatocyte paraffin-1 revealed that PFD enhanced liver regeneration. The research confirmed that PFD gradually downregulated elevated eIF6, P311, and TGF-β levels in BDL-induced LF. These findings suggest that PFD could be a potential treatment for LF, as it may help attenuate fibrosis and enhance liver regeneration, possibly through the modulation of these specific markers.
Introduction
Liver fibrosis (LF) is characterized by excessive extracellular matrix (ECM) production, primarily from activated hepatic stellate cells (HSCs). This process is exacerbated by the release of profibrotic cytokines such as transforming growth factor-beta (TGF-β1), which induces myofibroblast (MFB) differentiation and HSC activation (Breitkopf et al., 2006; Brown et al., 2006; Kisseleva and Brenner, 2021; Li et al., 2004; Meyer et al., 1990; Tan et al., 2021a; Win et al., 1993). Elevated TGF-β levels also lead to increased alpha-smooth muscle actin (α-SMA) levels, a marker for MFB (Derynck and Zhang, 2003; Tan et al., 2021a).
Eukaryotic translation initiation factor 6 (eIF6) plays a crucial role in hepatic steatosis and fibrogenesis, activating HSCs, promoting MFB differentiation, and advancing LF progression (Brina et al., 2015; Ding et al., 2015; Gallo and Manfrini, 2015; Renger and Wolstenholme, 1972; Scagliola et al., 2022; Spits and Di Santo, 2011; Valenzuela et al., 1982; Weis et al., 2015; Wurth and Gebauer, 2015; Zhang et al., 2014).
Recent research has shown that eIF6 interacts with neuronal protein 3.1 (P311, also known as NREP), inducing fibrogenesis through upregulation of TGF-β (Peng et al., 2012; Tan et al., 2010). P311 is highly upregulated in fibrosis-associated MFBs and is involved in ECM alteration and progression of various fibrosis through its positive effect on TGF-β1 expression (Lagares, 2019; Wang et al., 2010). Excessive P311 expression is associated with increased α-SMA and TGF-β1 levels through the TGF-β1/Smad signaling pathway. P311 itself activates HSCs through its effect on reactive oxygen species (ROS) and oxidative pathways.
P311 requires an interactive partner to obtain a stable tertiary structure and interacts with several cellular skeletal proteins, including nonmuscle myosin heavy chain 9, filamin A, eIF3b, and eIF6 (Stradiot et al., 2018). Previous research suggests that P311 and eIF6 may interact to influence ribosome synthesis, apoptosis, and the advancement of tissue fibrosis by directly affecting TGF-β1 and other downstream variables (Peng et al., 2012; Yue et al., 2014). Therefore, changes in the expression of the TGF-β factor through therapeutic drugs, eIF6, and P311 can lead to HSCs differentiation and MFB differentiation. The importance of examining P311 and eIF6 factors in this study lies in their high interaction with genes related to fibrosis and fibrosis-associated MFBs. Fibroblasts contribute in the ECM formation and deposition (Yang et al., 2015b). Drug therapy targeting the TGF-β gene can prevent fibrosis, by improving its expression of eIF6 and P311 factors, which reduce collagen accumulation and decrease HSC activation, and MFB differentiation, thus halting cirrhosis and HCC progression. These actions can overall prevent fibrosis and stop its progression.
Additionally, the pivotal role of HSCs, Kupffer cells, and macrophages are critically involved in the pathogenesis of LF. Kupffer cells, which are the resident macrophages of the liver, along with monocyte-derived macrophages, are essential in both the initiation and progression of fibrotic processes (Cheng et al., 2021). These cells secrete various pro-inflammatory and profibrotic cytokines, including TGF-β, which further stimulate HSC activation and enhance ECM deposition. Upon liver injury, Kupffer cells release ROS and other inflammatory mediators, thereby intensifying the fibrotic response (Ma et al., 2017).
Pirfenidone (5-methyl-1-phenyl-2(1H)-pyridone [PFD]) is a novel antifibrotic compound designed for treating idiopathic pulmonary fibrosis. It has been shown to reduce collagen deposition in various animal models in vivo (Iyer et al., 1999; Iyer et al., 1995; Sgalla et al., 2016; Shimizu et al., 1997; Shimizu et al., 1998). In vitro studies have demonstrated that PFD can prevent HSC activation and fibroblast proliferation in response to TGF-β-induced collagen synthesis (Li et al., 2021).
In this study, we assess the antifibrotic effects of PFD on experimental LF induced by bile duct ligation (BDL) in Wistar rats. Our aim is to provide further evidence supporting the application of PFD in managing experimental LF. Additionally, we investigate changes in the levels of eIF6 and P311 as related and multifunctional factors in LF. By exploring the behavior and function of eIF6 and P311 genes and downstream genes influenced by PFD, we suggest its potential as a novel therapeutic target in LF.
Materials and Methods
Animals
All animal treatments and handling procedures were approved by and conducted in accordance with the guidelines of the Animal Care and Handling Committee of Tarbiat Modares University (approval number: IR.MODARES.AEC.1402.032). Forty male Wistar rats (250 ± 30 g) were obtained from the Pasteur Institute of Iran. The animals were housed in stainless steel cages under controlled conditions (25 ± 2°C, 50% humidity, 12-h light/dark cycle) with daily cage cleaning. Rats had ad libitum access to clean tap water and standard rodent pellets throughout the study.
Experimental design
After a 1-week acclimation period, the 40 rats were divided into two main groups: Sham (n = 10) and BDL (n = 30). The BDL model was created through biliary obstruction, which involved abdominal surgery to ligate the common bile duct under ketamine-xylazine anesthesia following a 6-h fast, as described previously (Van Campenhout et al., 2019). The Sham group underwent the same surgical procedure without ligation. The BDL group was further divided into three subgroups (n = 10 each): BDL (untreated), BDL + PFD (200 mg/kg), BDL + PFD (500 mg/kg). The two treatment groups received oral PFD for 4 weeks, while the BDL and Sham groups were given saline. PFD doses were selected based on previous research (Garcıa et al., 2002). All rats were sacrificed on day 28 of the experiment.
Scarification, blood sample, and tissue collection
At the conclusion of the experiment, body weights were recorded. Rats were anesthetized with a ketamine/xylazine mixture, and blood samples were collected via cardiac puncture. Following exsanguination, the livers were removed and weighed to determine the liver index (liver weight/body weight ratio). Blood samples were centrifuged to obtain serum, which was stored at −80°C for further examination. The livers were divided into three sections: Fixed in 10% neutral buffered formalin for histological and immunohistochemical (IHC) studies, preserved in RNA later for RNA extraction, and snap-frozen in liquid nitrogen for biochemical and molecular studies. All experiments were conducted in triplicate using laboratory animals.
Biochemical and molecular studies
Biochemical parameters
Serum samples were applied for quantitative measurement of the biochemical markers, including alkaline phosphatase (ALP), aspartate aminotransferase (AST), and alanine aminotransferase (ALT). These were detected via enzymatic methods using the Pars Azmun Kit (Iran), following the manufacturer’s instructions.
Histological analysis
Liver samples were processed for histological analysis using standard techniques. Briefly, tissues were dehydrated, cleared, and embedded in paraffin blocks. Sections (5 μm thick) were cut using a microtome and mounted on glass slides. After deparaffinization, slides were stained with hematoxylin and eosin (H&E) for morphological analysis, Masson’s trichrome, and Sirius Red for collagen fiber content and liver structural changes. Staining was performed according to the manufacturers’ protocols (Sigma-Aldrich). The degree of fibrosis was assessed using Image J software. All samples were independently evaluated and scored by a pathologist using the fibrosis scoring system published by Gamal (Gamal and Khaled, 2011). Fibrosis severity was classified into seven stages (0–6), with 0 indicating no fibrosis and 6 denoting cirrhosis, based on the Ishak and METAVIR scoring systems (Ishak et al., 1995). The Ishak and METAVIR scoring systems for staging have been widely utilized, especially for LF, as shown in Table 1.
Scores for Liver Fibrosis Obtained by the Ishak and METAVIR Scoring Systems
IHC analyses
Liver sections from Wistar rats were immunohistochemically stained for hepatocyte paraffin-1 (HepPar-1) (Anti-Hepatocyte Specific Antigen antibody, mouse monoclonal antibody [OCH1E5], GeneTex, USA, dilution 1:100), α-SMA (Rabbit Anti-ACTA2 Polyclonal antibody, biorbyt, UK, dilution 1:500), and Ki-67, a proliferation marker (rabbit monoclonal antibody [SP6], BIOCARE MEDICAL, USA, dilution 1:50). Staining was performed using a Mouse and Rabbit Specific HRP/DAB Detection IHC kit (Abcam, UK, catalog number ab64264). After deparaffinization and hydration, sections underwent antigen retrieval and were incubated with primary and secondary antibodies. Stained sections were examined microscopically, and positive cells for each marker were counted across 12 consecutive fields to determine expression percentages, which were then analyzed with Image J software.
Hydroxyproline measurement
Hydroxyproline (Hyp) concentration in liver tissue was assessed using commercially available Hyp detection kits (Kiazist Life Sciences, Hamedan, Iran). The analysis was performed using colorimetry, following the manufacturer’s instructions.
RNA isolation and real-time PCR
Total RNA was isolated from the frozen liver tissues of different experimental groups using Trizol reagent (Yektatajhize, Iran) and stored at −80°C. One microgram of RNA was reverse-transcribed to complementary DNA (cDNA) by the Revert Aid First cDNA Synthesis Kit (Yektatajhize, Iran). For mRNA quantification, real-time PCR was performed to quantify the expression of eIF6, P311, TGF-β1, interleukin-10 (IL-10), tumor necrosis factor-alpha (TNF-α), IL-1, NF-κB, α-SMA, Col1a1, Acta2, Ccn2, Platelet-derived growth factor (PDGF), Vascular endothelial growth factor VEGF, matrix metalloproteinase-2 (MMP-2), and tissue inhibitors of metalloproteinase-1 (TIMP-1). Real Plus 2x Master Mix SYBR Green (Amplicon, Denmark) was used on an ABI-Step One system (Applied Biosystems, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression served as a reference. The sequences of oligonucleotide primer pairs used in RT-qPCR are detailed in Table 2, while Table 3 provides the temperature and cycle parameters. Relative quantification was calculated using the 2−ΔΔCt method.
Primer Pair Sequences Used for Real-Time PCR
α-SMA, alpha-smooth muscle actin; eIF6, eukaryotic translation initiation factor 6; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IL-1β, interleukin-1beta; MMP-2, matrix metalloproteinase-2; TIMP-1, tissue inhibitors of metalloproteinase; TGF-β, transforming growth factor beta; TNF-α, tumor necrosis factor-alpha.
Description of the Cycle and Temperature Applied in qPCR
Western blotting
Total proteins were extracted from frozen drug-treated and control liver tissue samples by homogenization in lysis buffer containing protease inhibitors. Protein content was measured using the bicinchoninic acid assay kit (Thermo Fisher Scientific, UK) as described in previous research (Yousefi et al., 2020). Equal amounts of protein sample (40 μg/lane) were loaded and separated using 10% SDS-PAGE, and then the proteins were transferred onto a PVDF membrane (Millipore, MA, USA). After blocking the membranes for 2 h with 5% w/v BSA in TBST solution, the membranes were incubated with primary antibodies against TGF-β1, eIF6, and P311 (1:1000) for an entire night at 4°C. GAPDH served as a loading control. Next, the membranes were exposed to Horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz, USA) for 60 min at room temperature (1:10,000). The protein bands on membranes were visualized by enhanced chemiluminescent substrate (Amersham, USA), and relative quantification of the proteins was performed using Image J software (National Institutes of Health, Bethesda, USA).
Statistical analysis
All data are presented as mean ± standard deviation. Statistical analysis was performed using GraphPad Prism version 9 (GraphPad Software Inc., San Diego, CA, USA). One-way analysis of variance was used to establish statistical significance, followed by Tukey’s post hoc test for multiple comparisons. The threshold for statistical significance was set at p < 0.05. All experiments were conducted in triplicate using laboratory animals.
Results
Effects of PFD on serum levels of biochemical parameters and liver hydroxyproline content
Figure 1A illustrates the experimental design. BDL significantly elevated serum levels of liver enzymes ALT, AST, and ALP (Fig. 1B). Both doses of PFD significantly decreased these enzyme levels compared to the BDL group. Hydroxyproline (Hyp), a characteristic component of collagen, was significantly higher in the BDL group compared to the sham group. PFD treatments significantly lowered Hyp content compared to the BDL group, indicating reduced collagen deposition.
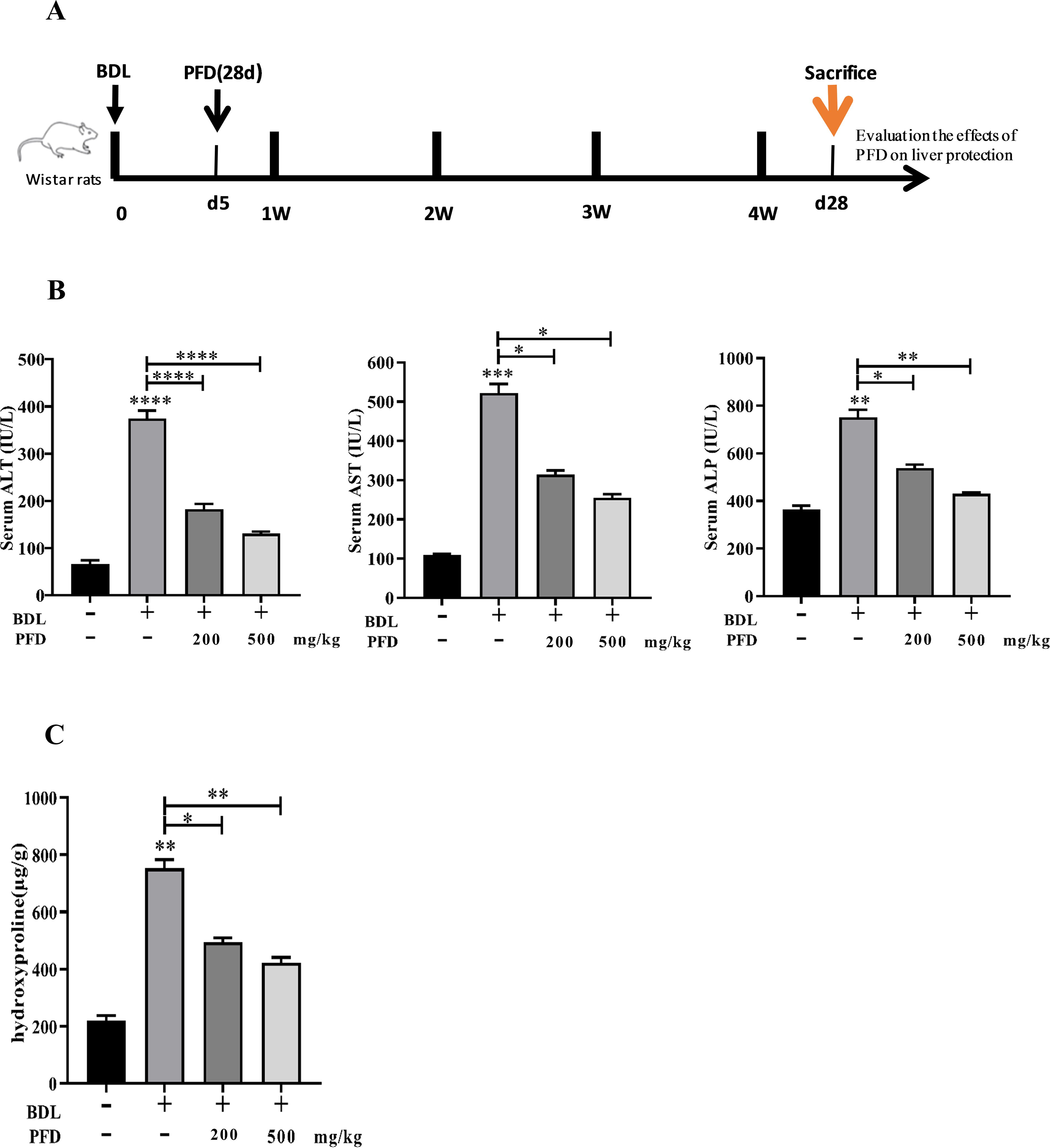
The effects of pirfenidone on biochemical parameters and Hyp. After 28 days of either BDL or sham surgery, Wistar rats were sacrificed.
PFD alleviates BDL-induced LF
The BDL rat model was created to explore PFD’s effects on LF. As shown in Fig. 2A, B DL caused macroscopically evident changes. Daily administration of 200 and 500 mg/kg PFD improved gross nodular appearance and liver texture, with relatively reduced fibrosis. Histopathological examination of liver sections (H&E, Sirius Red, and Masson trichrome staining) revealed that the structure of the hepatic lobule was clear in the sham group, while the BDL group showed significant collagen deposition, bile duct proliferation along with infiltration of inflammatory cells. However, the findings indicated that PFD reduced collagen deposition and necrosis surrounding the portal tract in BDL groups, and a dose–response correlation was revealed. Furthermore, PFD markedly decreased fibrotic score and fibrosis area percentage compared to the BDL group (Fig. 2B and C). In addition, in the BDL group, livers had become substantially enlarged and had lost compliance when compared to rats who were given a sham operation. BDL significantly increased the liver/body weight ratio compared with sham-operated rats. The body weight/liver ratio changed considerably less in the BDL+PFD groups than in the other groups (Fig. 2D).
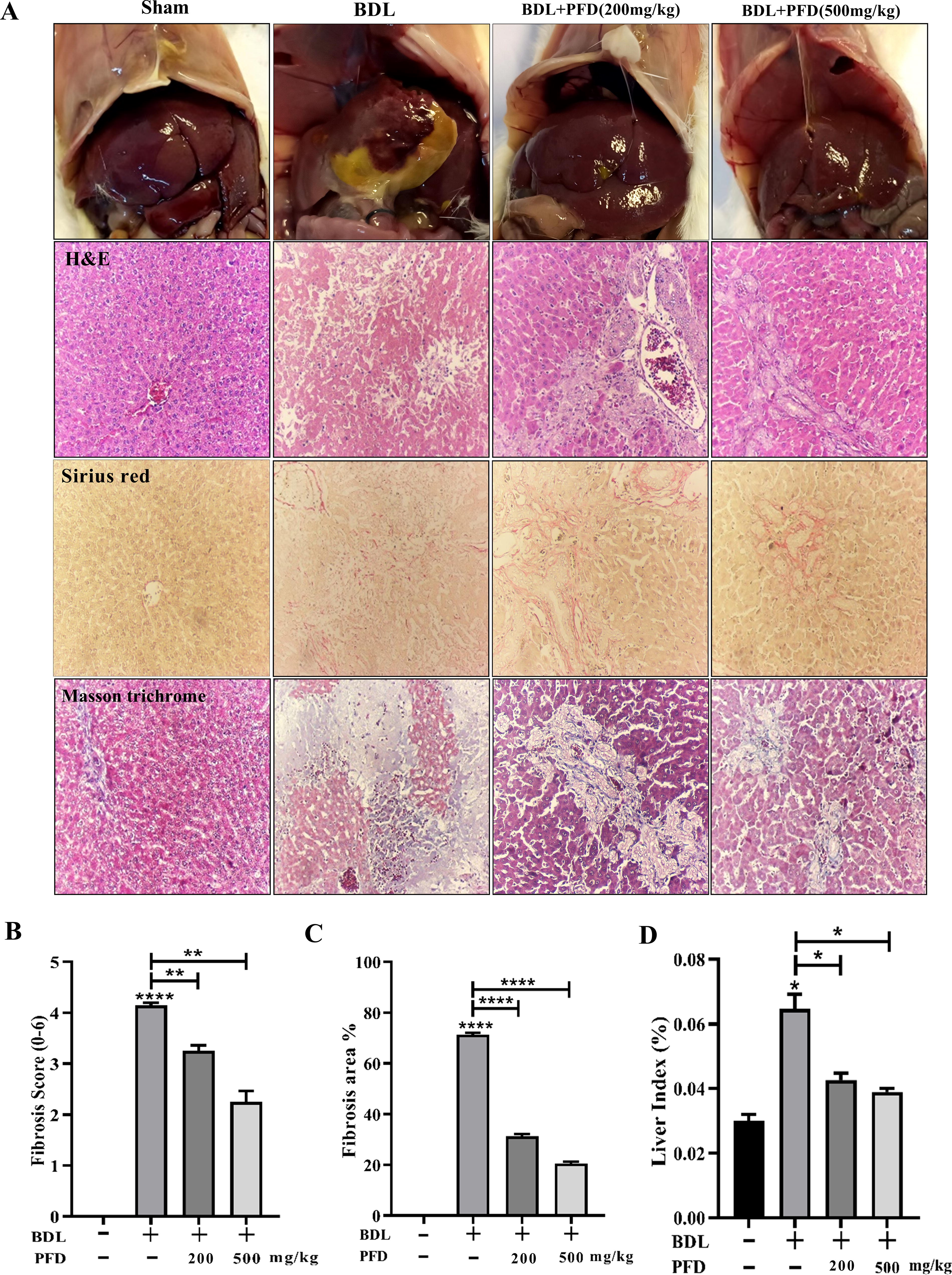
Pirfenidone improves LF induced by BDL.
PFD decreases HSC activation in BDL-induced LF
HSCs are the main ECM-producing cells in LF, activated by TGF-β1 release. We examined α-SMA expression using IHC labeling to determine PFD’s effect on collagen synthesis reduction. A unique marker for activated HSCs seems to be α-SMA. Immunostaining found a slight amount of α-SMA expression on the bile ducts and peripheral vascular in the sham group. BDL-induced LF was further confirmed by the considerable increase in α-SMA mRNA expression percentage that showed the activation of HSCs during LF. In addition, upon administration of PFD, the expression of α-SMA was gradually downregulated around the portal area with rising doses of PFD in BDL-induced groups, advocating that PFD suppresses HSC activation compared to the BDL group (Fig. 3A–C).
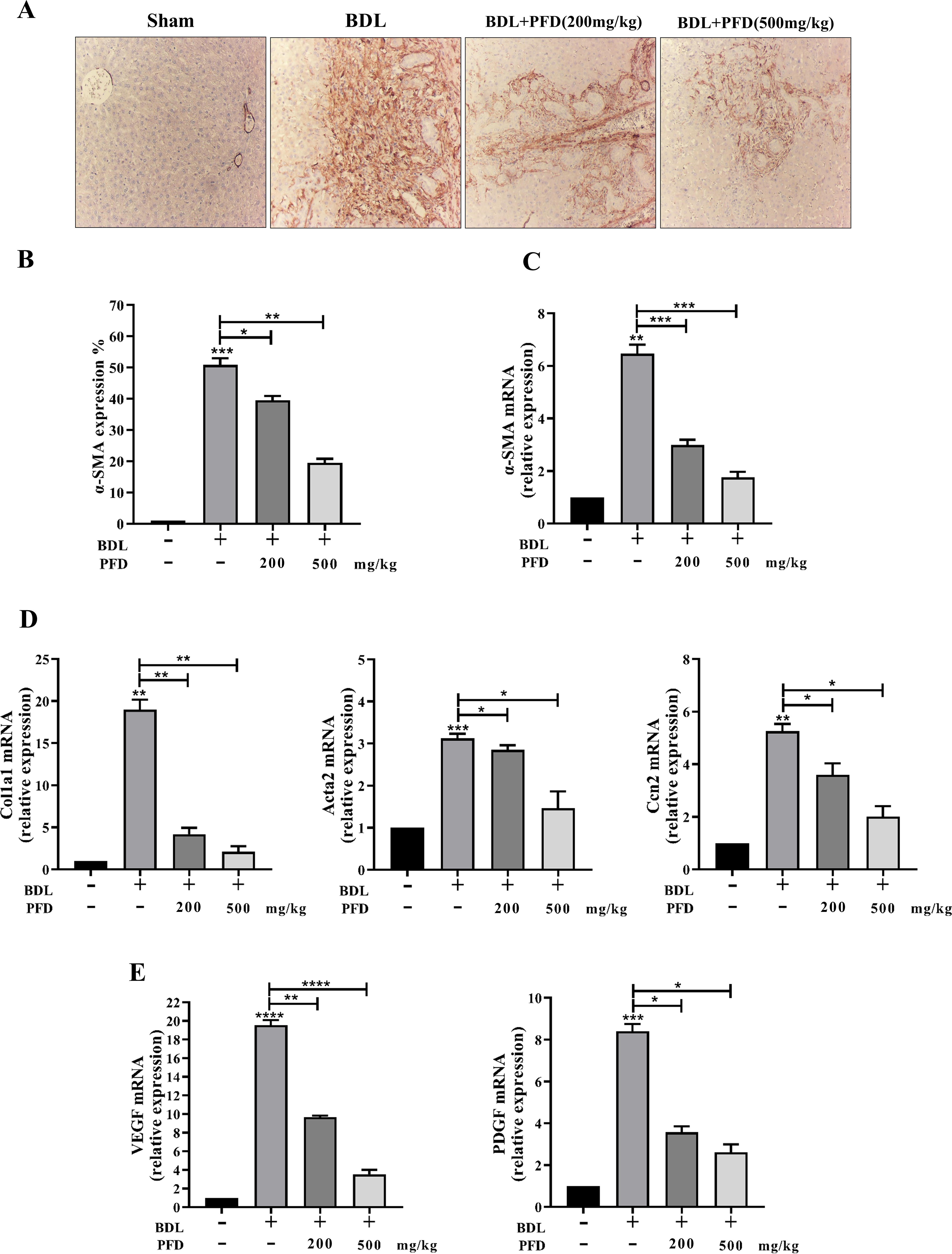
PFD decreased HSC activation in BDL-induced LF.
Effect of PFD on
RT-PCR to quantitatively examine the expression of genes that are believed to be crucial for the accumulation of ECM. After PFD treatment, there was a noticeable suppression of the RNA expression of fibrogenesis genes, such as Acta2 (encoded by α-SMA), Cola1, and Ccn2, as compared to the model group. In BDL-induced LF, PFD treatment resulted in dose-dependent upregulation of collagen in fibrotic livers (Fig. 3D). In accordance with these findings, we used RT-PCR to examine VEGF and PDGF expression was performed in order to more corroborate the impact of PFD on HSC activation. VEGF and PDGF are strong, proliferative, and active cytokines in the development of HSCs. This calculation showed that PFD dramatically reduces PDGF and VEGF expression levels (Fig. 3E). These findings imply that PFD may mitigate HSC activation, which would lessen the accumulation of ECM.
Effects of PFD on ECM remodeling in BDL-induced LF
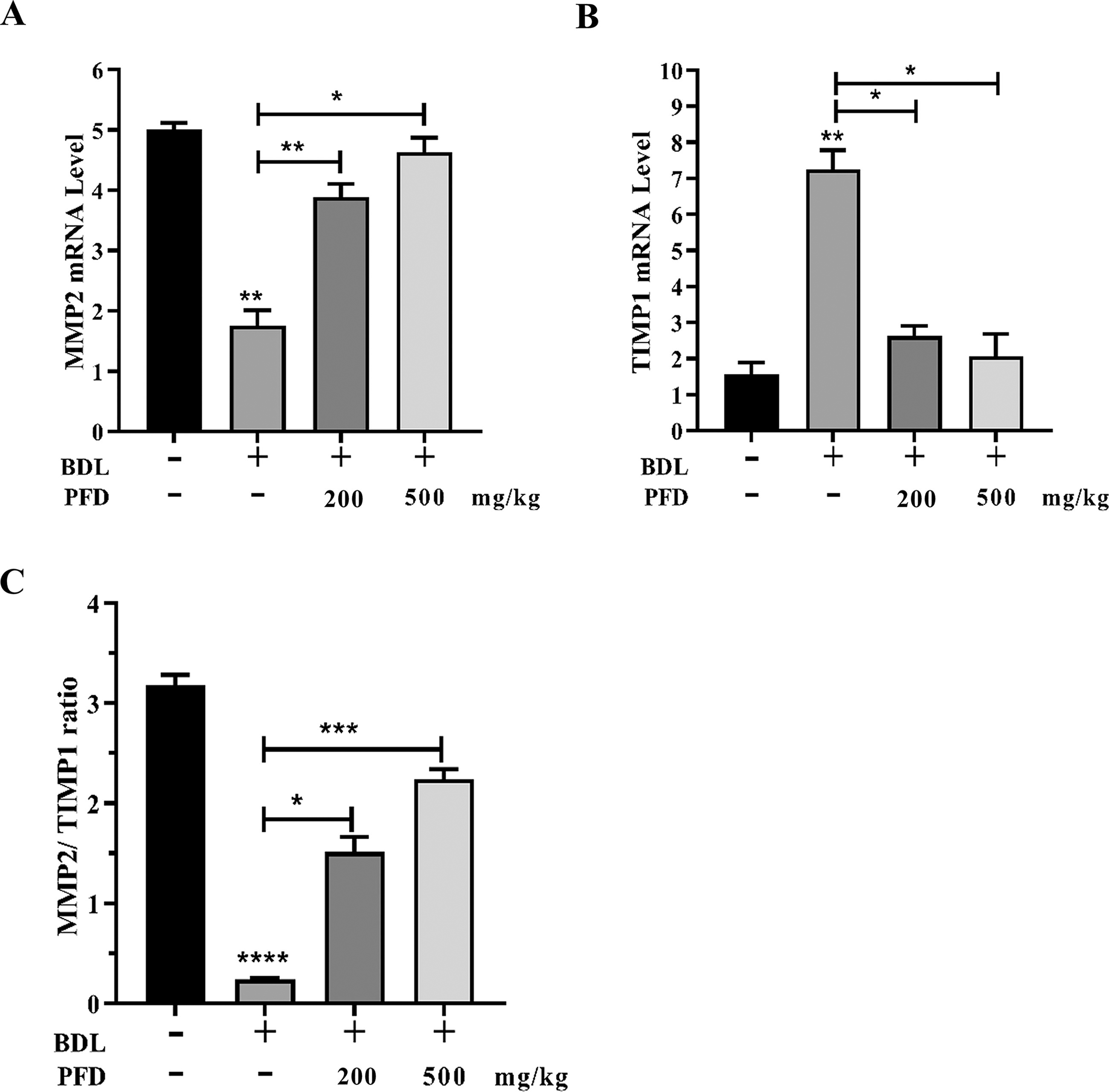
LF is primarily caused by an imbalance between ECM degradation and synthesis. MMPs and TIMPs are key regulators of ECM metabolism in the liver. TIMPs inhibit MMPs and stop ECM degradation. These reactions also promote LF. Our investigation revealed that PFD treatment significantly enhanced MMP-2 expression in BDL-induced LF. The TIMP1 gene was strongly repressed following PFD administration. The BDL group exhibits a preference for liver fibrogenesis over fibrolysis, as seen by the notable upregulation of TIMP-1 and downregulation of MMP-2 gene expression, which ultimately leads to a reduction of the MMP-2/TIMP-1 ratio in comparison to normal rats. After PFD was administered, there was a dose-dependent shift in the balance toward the upregulation of MMP-2 gene expression and the downregulation of TIMP-1, with an elevated MMP-2/TIMP-1 ratio (p < 0.05) (Fig. 4A,B and C). This may help the LF regression process. The active form of TGF-β1 enhances the synthesis of numerous ECM proteins and downregulates MMPs’ destruction of those proteins through the activation of TIMPs in HSC/MFB. These results suggest that PFD might reduce ECM accumulations brought on by LF brought on by BDL.
Effects of PFD on pro-inflammatory mediators in BDL-induced LF
Inflammation is a significant characteristic of LF, promoting the survival of activated HSCs. To examine the impact of PFD on the inflammatory response, we quantitatively detected the mRNA expression of IL-1, IL-10, TNF-α, and NF-κB in BDL-induced liver injury using RT-PCR. The results showed that BDL-induced LF led to a significant elevation in hepatic IL-1, NF-κB, and TNF-α levels and a decrease in IL-10 compared with the sham group. PFD treatment reversed these changes in a dose-dependent manner compared with the model group (Fig. 5A–D). These findings suggest that PFD reduced hepatic inflammatory infiltration in BDL rats.
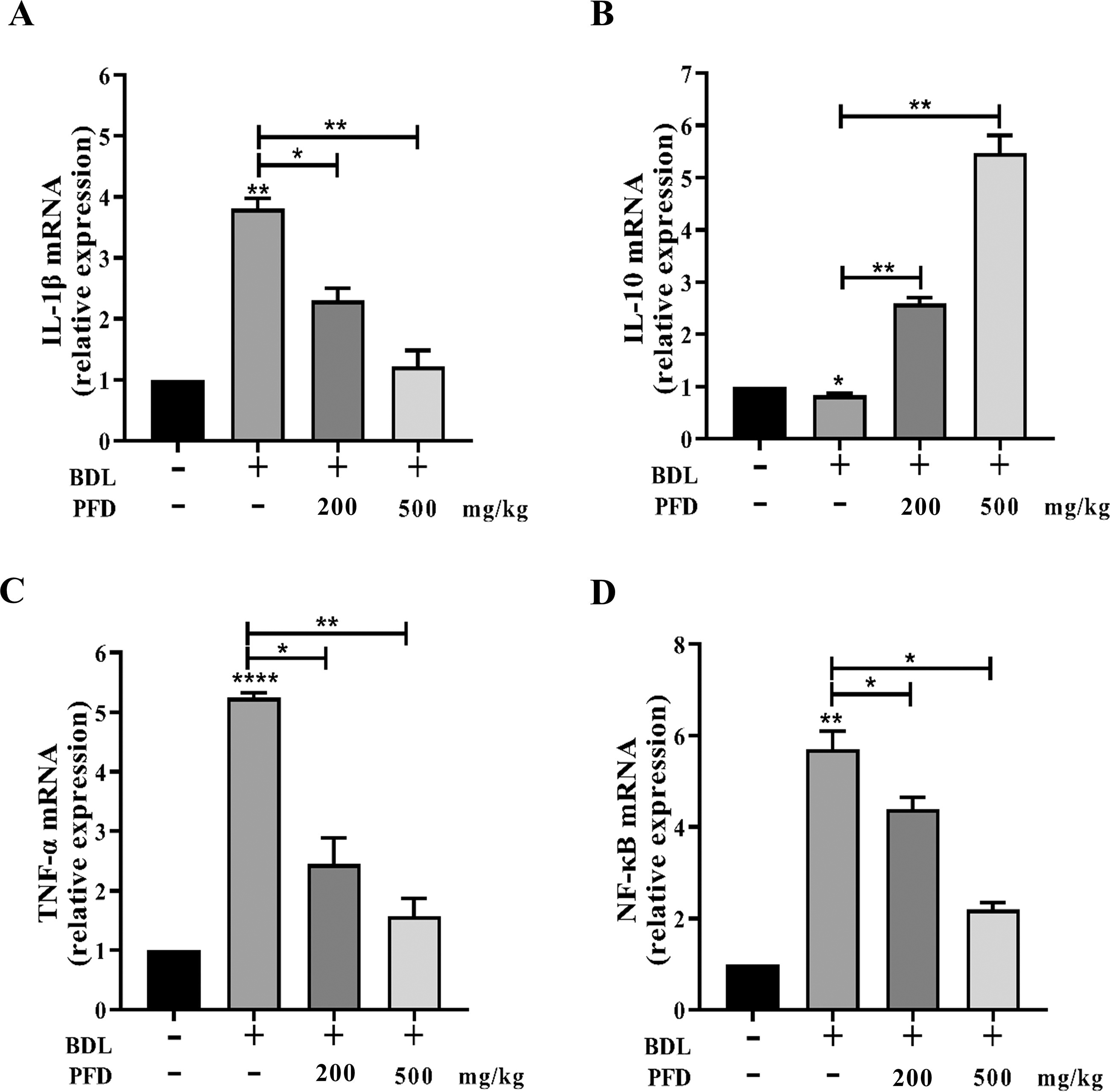
The effects of PFD on the inflammatory response. The mRNA levels of
Effects of PFD on eIf6, P311, and TGF-β protein levels in BDL-induced LF rats
Western blotting was used to examine the staining intensities of eIF6, P311, and TGF-β proteins in rats with BDL-induced hepatic fibrogenesis. The findings showed that in model group eIF6, P311, and TGF-β expression levels were purposefully enhanced compared to the sham group (Fig. 6A and B). However, the protein expression levels of eIF6, P311, and TGF-β were gradually reduced with increasing doses of PFD in the BDL-induced LF rats compared with the BDL group. The RT-qPCR analysis results for eIF6, P311, and TGF-β were consistent with western blotting findings (Fig. 6C). These results suggest that PFD may affect eIF6 and P311 in BDL rats, potentially leading to mitigated LF.

Effects of PFD on HepPar-1 and Ki-67 immunostaining
HepPar-1 is a monoclonal antibody that reacts with an epitope on hepatocyte mitochondria membranes, exhibiting a characteristic granular cytoplasmic pattern. It is regarded as the most sensitive and specific positive marker of hepatocellular differentiation (Lamps and Folpe, 2003). High HepPar-1 expression in hepatocellular carcinoma is considered a favorable prognostic factor (Mondada et al., 2006). To evaluate the impact of PFD on HepPar-1 expression, immunohistochemistry was used on a variety of liver tissues from the experimental groups. Representative photomicrographs of IHC staining of HepPar-1 and Ki-67 are presented in Figure 7A. The control animal’s liver sections displayed a characteristic dense granular cytoplasmic response. However, the BDL group showed a weak response and a heterogeneous distribution in the majority of hepatocytes, indicating that BDL had a negative effect on HepPar-1 expression. The majority of hepatocytes in PFD groups exhibited an increasingly moderate immune response. The majority of hepatocytes in liver sections taken from BDL+PFD (500 mg/kg) rats showed a dense granular cytoplasmic HepPar-1 immunological response (Fig. 7B). To confirm the previous findings, histomorphometric examination of the liver sections with Ki-67 immunostaining was carried out, in which a mildly positive nuclear Ki-67 was noticed in BDL sections compared to the control group. Meanwhile, either dosage of PFD supplementation significantly increased the expression of Ki-67 compared to the BDL group, suggesting its favorable effects on Ki-67 expression.
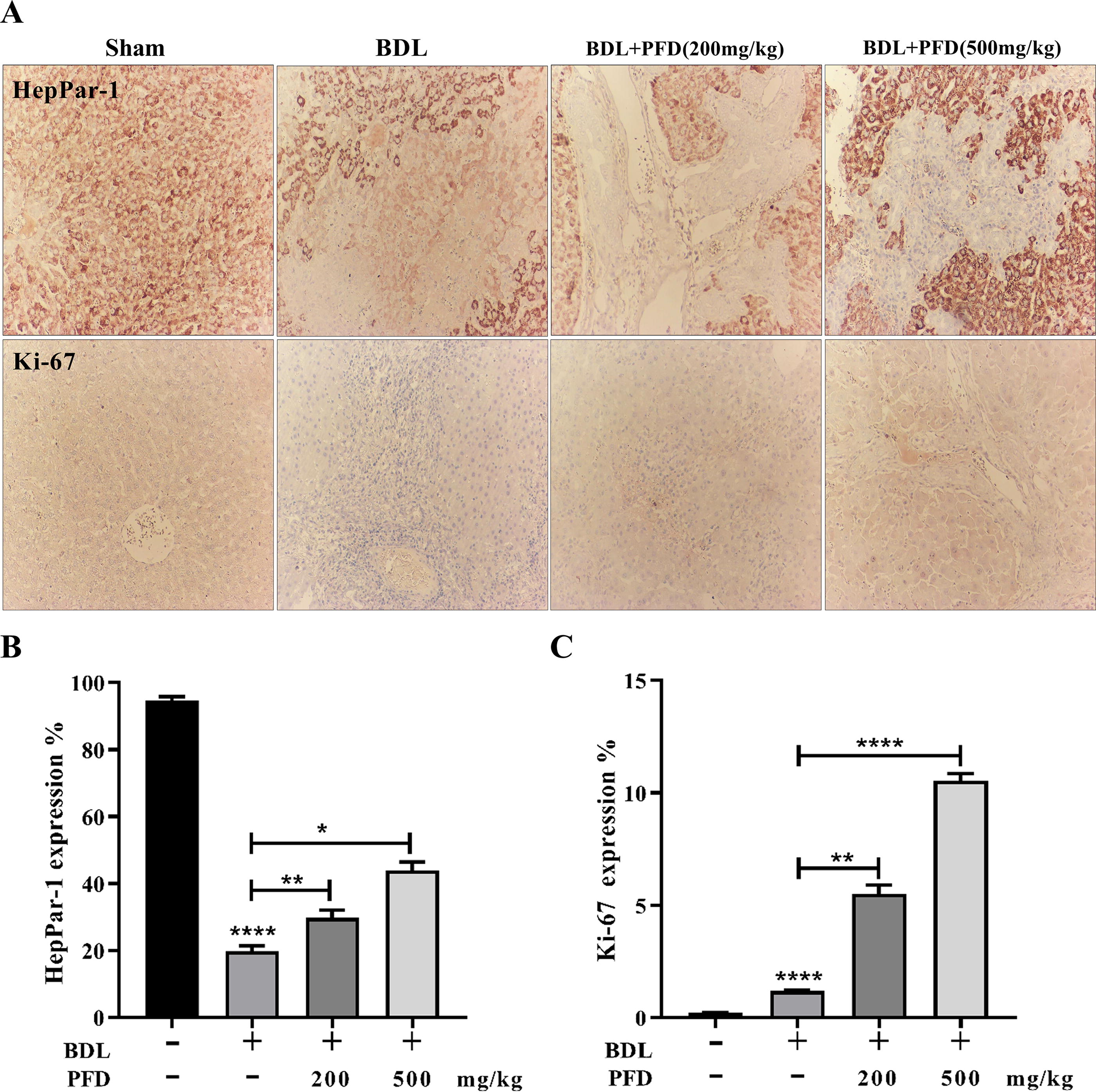
Effects of pfd on immunohistochemical stained sections of heppar-1 and ki-67in bdl-induced wistar rats.
Discussion
Liver tissue exhibits a remarkable capacity for regeneration following injury (Erdelmeier et al., 1998). However, prolonged exposure to hepatic stress without treatment impairs this regenerative ability and induces LF. This process is characterized by a significant reduction in hepatocyte proliferation, ultimately leading to organ failure and cirrhosis (Gilgenkrantz and Collin del’Hortet, 2011). While liver transplantation is considered the ideal treatment for cirrhosis, the scarcity of available donors makes it inaccessible for most patients. Consequently, there is an urgent need for alternative treatments, such as repurposing antifibrotic drugs, that can stimulate liver regeneration.
BDL-induced LF is unique in that the bile duct epithelium is the focal point of most pathological lesions. The BDL process subjects the biliary epithelium to biomechanical stress, initially causing biliary epithelial cells (BECs) to proliferate and expand. This pathological change is termed “bile duct reaction” or “reactive bile duct epithelial cells” (Choi and Diehl, 2009). Proliferating BECs can induce cholestatic LF by stimulating the proliferation and activation of MFBs, Kupffer cells, and HSCs leading to ECM production and liver damage (Matsumoto et al., 1994; Sato et al., 2016). The ductular reaction has several detrimental effects including suppression of liver regeneration, exacerbation of inflammation, and acceleration of LF (Nuñez-Garcia et al., 2017; Syn et al., 2011; Wang et al., 2014; Whitington et al., 2005; Yang et al., 2014). Previous research has shown that inhibiting the ductular response can reduce inflammation, cholestasis-induced liver damage, and fibrosis (Coombes et al., 2015; Wang et al., 2017).
Understanding the development of fibrosis remains one of the greatest challenges for researchers (Amar and Bianca, 2016; Hu and Phan, 2016; Meng et al., 2016). Despite notable progress, managing fibrosis continues to be a difficult task (Pu et al., 2013; Richeldi, 2016; Wasmuth, 2010). Clinical experience has demonstrated that PFD significantly reduces fibrosis, although its exact function in LF remains uncertain.
PFD, an effective antifibrotic drug, exerts anti-inflammatory, HSC activation inhibition, ECM deposition regulation, and regenerative properties. These effects have been observed in several fibrosis models, with histopathological and biochemical impacts (Liu et al., 2017a; Liu et al., 2017b; Pourgholamhossein et al., 2018). Studies have shown that PFD dramatically ameliorates the extent of fibrosis in the kidney, lung, and liver of various animal models. A recent study demonstrated that PFD could ameliorate liver injury, although its mechanisms require further elucidation (Xi et al., 2021). The strong therapeutic value of PFD in LF, regardless of the diverse pathogenic causes, suggests that it may target the main signaling pathways associated with LF development. In the present study, we focused on the preventive effects of PFD on LF, as preventing the development of fibrosis is generally more feasible than reversing established advanced fibrosis.
The present study demonstrates that PFD at doses of 200 and 500 mg/kg significantly ameliorates bile duct proliferation and exerts remarkable antifibrotic and anti-inflammatory effects, promoting greater liver regeneration in BDL-induced LF. These effects were confirmed through histopathological analysis (hepatic architecture recovery), immunohistochemistry, biochemical improvements, and quantitative analysis of HSC activation, inflammatory response, and ECM deposition-related genes. Histological staining (H&E, Masson’s Trichrome, and Sirius Red) showed that PFD significantly decreased the fibrosis score and area in BDL-induced LF by approximately 40% and 50% at doses of 200 and 500 mg/kg, respectively. These findings align with previous studies: Tada et al. reported a 40% reduction in dimethylnitrosamine-induced LF with PFD treatment (Tada et al., 2001), while Garcia et al. observed a 50% reduction in BDL-induced LF and a 40–70% reduction in CCl4-induced LF (Garcıa et al., 2002). Recent studies by Antar et al. and Xie et al. suggest multiple mechanisms of action for PFD, highlighting its promise in treating inflammation and fibrosis, including NAFLD (Antar et al., 2022; Xie et al., 2023). Additionally, earlier research indicates that PFD can improve tissue fibrosis by reducing HSC activation, modulating biochemical factors, and decreasing ECM deposition (Chen et al., 2019; Lv et al., 2020; Poo et al., 2020; Seniutkin et al., 2018). The outcomes of the current study corroborate these prior investigations and provide additional evidence of PFD’s antifibrotic properties in animal models of fibrosis.
Our study also revealed PFD’s potential for promoting liver regeneration, as evidenced by notable alterations in VEGF and PDGF expression. These changes were accompanied by increased HepPar1 and Ki-67 immunostaining, which are markers of functioning hepatocytes (Dubuquoy et al., 2015). In line with previous findings (Bingül et al., 2016; Lee et al., 2013; Mohamad et al., 2022), our results demonstrate that PFD mitigates BDL-induced liver injury by significantly lowering transaminases, improving the liver index, and enhancing HepPar-1 immunostaining. These effects collectively indicate improved hepatic function. Alongside these improvements, we also observed enhancements in hepatic architecture. While BDL-group rats displayed mild Ki-67 immunostaining, suggesting hepatocyte proliferation as part of the adaptive response to liver tissue damage (Ghanim et al., 2021; Kuramitsu et al., 2013; Piekarska et al., 2006), PFD treatment prompted strong, diffuse nuclear Ki-67 immunostaining. This observation suggests that PFD may induce a more pronounced increase in hepatocyte proliferation, potentially leading to more effective liver regeneration.
Kupffer cells first create damage and start the inflammatory cascade reaction in liver injury. They then release a number of inflammatory factors and draw macrophages to the injury site, which exacerbates the inflammatory injury. By releasing pro-inflammatory and profibrotic mediators, the paracrine effects of macrophages and Kupffer cells can influence HSC activation and sustain its survival, aggravating the inflammatory response and ensuring HSCs continue to activate and survive (Tan et al., 2021a). Small molecule inhibitors can stop macrophage invasion by suppressing the Wnt/β-catenin signaling pathway in HSCs. This leads to significantly lower HSC activation, less hepatic inflammation, and less ECM accumulation in a mouse model of CCl4-induced LF (Akcora et al., 2018). Activated HSCs, as the primary producers of ECM, play a crucial role in liver fibrogenesis. Key antifibrotic activities are indicated by factors controlling ECM production and HSC induction, proliferation, and function. MFBs can be further activated by excessive cytokines, leading to substantial ECM aggregation and deposition (Wang et al., 2022). Abnormal ECM deposition results in aberrant wound healing responses, which in turn promote the progression of LF (Ding et al., 2020; Zhao et al., 2021). Research has shown that liver sinusoidal endothelial cells (LSECs) can trigger HSC activation. Fibrotic growth factors, particularly TGF-β1 and PDGF, have been demonstrated to be crucial for LSECs in supporting HSC activation (Lee et al., 2007). Understanding these mechanisms provides insight into the complex interplay of cellular and molecular factors involved in LF and regeneration.
To assess the impact of PFD on HSCs, we analyzed the expression of VEGF and PDGF genes, along with the α-SMA marker. It is widely accepted that various growth factors play crucial roles in regulating liver regeneration. VEGF, in particular, is closely associated with this process and significantly contributes to LF (Marrone et al., 2016). Notably, increased VEGF expression has been shown to enhance hepatocyte proliferation during liver regeneration (Wang et al., 2012). Moreover, VEGF stimulates the proliferation of sinusoidal endothelial cells in the liver, essential for effective regeneration (DeLeve et al., 2008). As a major angiogenic agent, VEGF promotes neovascularization and improves the trophic portal blood supply to proliferating hepatocytes, supporting the liver regeneration process (Bockhorn et al., 2007; Dirscherl et al., 2019). PDGF, on the contrary, may accelerate LF by increasing VEGF expression and matrix protein synthesis in HSCs (with Kupffer cells accompanied) (Hammerich and Tacke, 2023; Thabut et al., 2011; Zhang et al., 2014). It is the most potent stimulator of HSC proliferation, chemotaxis, and phenotypic transition to MFBs, primarily driving PDGF-mediated intracellular signaling that promotes collagen synthesis (Roehlen et al., 2020; Thabut et al., 2011).
In liver tissue from patients with chronic liver diseases, the extent of fibrosis appears to correlate closely with PDGF expression (Zhou et al., 2016). Interestingly, transgenic mice overexpressing PDGF developed LF independently of increased TGF-β expression (Czochra et al., 2006). This functional understanding of PDGF and VEGF signaling has sparked a growing interest in antifibrotic drugs targeting these pathways. Given that PDGF is known to modify HSC-based vascular development, causing HSCs to acquire an angiogenic phenotype, these findings suggest that both VEGF signaling in sinusoidal endothelial cells and PDGF signaling blockade in HSCs contribute to the antiangiogenic effects of antifibrotic medications (Aleffi et al., 2011; Hayes et al., 2014; Kocabayoglu et al., 2015; Zhang et al., 2014). These results underscore the functional relationship between LF development and intrahepatic angiogenesis.
In this study, we demonstrated that PFD dose-dependently reduced the expression of collagen deposition marker mRNA and α-SMA in fibrotic tissues. These findings suggest that the suppression of HSC activation and the rebalancing of ECM deposition are potential molecular mechanisms underlying PFD’s antifibrotic action.
Our data revealed that BDL-induced rats exhibited excessive collagen deposition, as detected by Masson’s trichrome staining. We analyzed Col1a1, Acta2, Ccn2, and the MMP-2/TIMP1 ratio to assess ECM deposition. We observed a significant reduction in expression at both doses of PFD in BDL-induced LF. This finding indicates that PFD attenuates LF, at least partially, by reducing HSC proliferation and activation, as well as remodeling the ECM.
A unique aspect of this study was the evaluation of eIF6 and P311 alterations alongside TGF-β in different study groups. We developed an in vivo model of LF using BDL to assess eIF6, P311, and TGF-β regulation. Our results showed that BDL-induced liver injury significantly increased the expression levels of eIF6, P311, and TGF-β. Conversely, administration of either dose of PFD significantly reduced eIF6, P311, and TGF-β gene expression. Research has shown that eIF6, which controls the binding of 40S and 60S ribosomal subunits to promote ribosomal biogenesis and translational regulation, regulates fibrosis via the TGF-β pathway in animal models (Yang et al., 2015b), human hypertrophic scarring (Yang et al., 2015a), and chronic kidney disease (Yao et al., 2015). eIF6 also controls the MMP-2/TIMP-2 ratios to maintain a balance between ECM deposition and degradation (Bai et al., 2016). Furthermore, the results indicate that eIF6 may regulate Sp1 recruitment to the TGF-β1 promoter via H2A.Z occupancy during fibrogenesis, thus influencing TGF-β (Yang et al., 2015b).
Previous research indicates that eIF6-regulated MFB activation and differentiation are mediated through the TGF-β signaling pathway (Shu et al., 2016). P311, a protein, was found to be the most highly upregulated gene across multiple fibroses due to its favorable impact on TGF-β1 expression. Guimaraes et al. reported that P311 is expressed in HSCs. P311 expression showed a five-fold increase in gene expression following in vitro and in vivo activation (both during CCl4 and BDL-induced LF) in a major microarray study comparing gene expression levels in activated HSCs (Guimarães et al., 2015). Interestingly, an early study using metallothionein-deficient mutant and wild-type SV-40 immortalized HSCs revealed that this protein regulates P311 expression in HSCs (Miura and Naganuma, 2000). It’s worth noting that research shows TGF-β can trigger P311 production in vitro (Leung et al., 2008; Paliwal et al., 2004) and that P311 can modify TGF-β signaling (Guimarães et al., 2015). It has been demonstrated that P311 gene expression increases after HSC activation in vitro and in vivo and induces LF by activating TGF-β (Duan et al., 2019; Guimarães et al., 2015; Li et al., 2016).
Peng et al. demonstrated through immunohistological analysis that eIF6 and P311 overexpression were strongly colocalized in tissue. Evidence of the association between P311 and eIF6 was obtained through FRET in lung adenocarcinomas and co-immunoprecipitation experiments in HEK293 cells. Collectively, these findings indicate that eIF6 is a novel partner of P311 in certain tissues (Peng et al., 2012). Further observations suggest that PFD may reduce TGF-β activation via eIF6 and P311. Notably, eIF6 and P311 have been shown to potentially interact and influence TGF-β modulation in fibrosis (Peng et al., 2012; Tan et al., 2010). It can be concluded that TGF-β, eIF6, and P311 could contribute to mitigating BDL-induced LF through PFD’s influence. We anticipate that modulation of these factors will lead to significant physiological effects.
Conclusion
The study’s findings indicate that PFD treatment histologically protects liver tissue from the damaging effects of BDL. It enhances regenerative response and reduces inflammation, HSC activation, and ECM accumulation, thereby mitigating LF. All molecular and biochemical outcomes have been further corroborated through histopathological and IHC findings. This evidence suggests that PFD may be a promising therapeutic intervention for LF. However, clinical trials are necessary to evaluate the efficacy and safety of PFD in humans. Moreover, PFD alleviates eIF6, P311, and TGF-β1 expression. We speculate that the expression of these genes may mediate the hepatoprotective effect of PFD. However, the interplay between eIF6, P311, and TGF-β1 and their role in LF requires further in-depth studies. Our findings have uncovered a novel potential mechanism for the purported hepatoprotective effect of PFD via eIF6 and P311 expression modification, which may have implications for LF treatment.
Footnotes
Acknowledgment
The authors would like to express their gratitude to all laboratory management staff of the Tarbiat Modares University for the laboratory equipment provided. The authors would like to acknowledge the financial support of Modares Science and Technology Park for this project.
Authors’ Contributions
Z.Y.: investigation, writing original draft and editing, formal analysis, and methodology; A.S.L.: supervision, investigation, editing, funding acquisition, validation, visualization and project administration; M.N.: supervision, validation, visualization and funding acquisition.
Disclaimer
The authors listed on the article are not employed by a government agency that has a primary function other than research and/or education.
Author Disclosure Statement
The authors declare no conflicts of interest, financial or otherwise.