
Abstract
High-throughput DNA sequencing has accelerated the discovery of disease-causing genetic variants, yet only in 10–40% of cases yield a genetic diagnosis. Increased implementation of genome sequencing has enabled a deeper exploration of the noncoding genome and recognition of noncoding variants as major contributors to disease. In a recent study, we identified a deep intronic variant in the AutoImmune REgulator (AIRE) gene (c.1504–818 G>A) as the cause of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), a life-threatening monogenic autoimmune disorder most often caused by biallelic AIRE defects. This deep intronic variant disrupts normal splicing AIRE , causing pseudoexon inclusion and altered protein function. By developing an antisense oligonucleotide (ASO) targeting the pseudoexon sequence, we restored normal AIRE transcript in vitro, thereby revealing a potential genotype-specific candidate treatment. Our study illustrates key aspects of intronic variant detection, validation, and candidate ASO development. Herein, we briefly highlight the growing potential of ASO-based therapies for deep intronic variants, addressing the unmet need of personalized, genotype-specific therapies in diseases lacking curative options.
High-throughput DNA sequencing has become increasingly affordable and accessible, accelerating the discovery of disease-causing variants. Despite these advances, a genetic cause is identified in only 10–40% of cases (Stranneheim et al., 2021; Thaventhiran et al., 2020). The search for causal variants has primarily focused on the coding genome, where most disease-causing variants have been discovered to date. Large-scale exploration of the noncoding genome, which accounts for 98% of our DNA and encompasses introns, promoter regions, untranslated regions, noncoding RNA, and intergenic regions, has become more widely accessible during the past decade. This is largely due to reductions in computing and storage costs, the availability of large genome sequencing (GS) databases, and novel bioinformatic tools (Wojcik et al., 2023). In the coming years, we can expect a further utilization of GS and a significant increase in the detection of disease-causing noncoding variants, carrying important diagnostic and therapeutic implications.
In our recent study, we identified a deep intronic variant as a recurring cause of APECED (autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy) (Ochoa et al., 2024), a monogenic autoimmune disease most often caused by biallelic deleterious variants in the AutoImmune REgulator (AIRE) gene (Constantine and Lionakis, 2019). AIRE promotes negative selection, a process by which self-reactive thymocytes are eliminated before exiting the thymus (Anderson et al., 2002). When AIRE function is impaired, self-reactive T cells escape to the periphery and cause early-onset autoimmunity in endocrine and non-endocrine organs. APECED is a life-threatening disease, with most patients developing multiple autoimmune complications, and confers a high morbidity and mortality (Constantine and Lionakis, 2019).
Over the course of 10 years, we identified 14 families (17 patients) showing typical clinical manifestations of APECED but without biallelic deleterious variants in exons or flanking intronic regions of AIRE. Most patients (15/17) were of Puerto Rican ancestry. Initial analyses by exome sequencing identified a common synonymous variant in AIRE (chr21:45709568 C>T, p.G227), which co-segregated with the disease, suggesting linkage to a potential noncoding causal variant within a shared haplotype. Further analyses confirmed the presence of a shared haplotype and a single overlapping region of absence of heterozygosity (AOH) on chromosome 21, encompassing the AIRE gene locus. Filtering for rare homozygous noncoding variants in this region uncovered a candidate deep intronic variant in intron 12 (AIRE c.1504-818 G>A). Subsequent GS analyses revealed that AIRE c.1504-818 G>A correctly segregated with the disease in all 14 families and was flagged by several splicing predictor tools as a potential cryptic splice site.
We hypothesized that this variant disrupts AIRE function through abnormal splicing. However, as AIRE is primarily expressed in the thymus, studying its transcript in primary patient cells posed a major challenge. To overcome this, we developed an extra-thymic AIRE-expressing cellular system using patient monocyte-derived dendritic cells (moDCs), which we stimulated with CD40 and receptor activator of nuclear factor-κB ligand (RANKL), two key signals necessary for AIRE expression in the thymus. Evaluation of AIRE transcript in moDCs revealed that AIRE c.1504-818 G>A causes the inclusion of an out-of-frame 109-base pair pseudoexon in all 17 patients. Protein modeling and transcriptomic analyses of AIRE-transfected HEK293 and thymic epithelial cells (TEC4D6) showed that this variant alters the C-terminus of the protein (Fig. 1) and disrupts its function. Notably, AIRE c.1504-818 G>A accounted for ∼10–15% of our NIH APECED cohort, is present in ∼0.1% of Latino/Hispanic individuals in gnomAD and was responsible for APECED in all our patients of Puerto Rican descent. The discovery of this variant in two individuals without Puerto Rican ancestry (one Spanish, one of mixed European ancestry) suggests a broader geographic distribution.
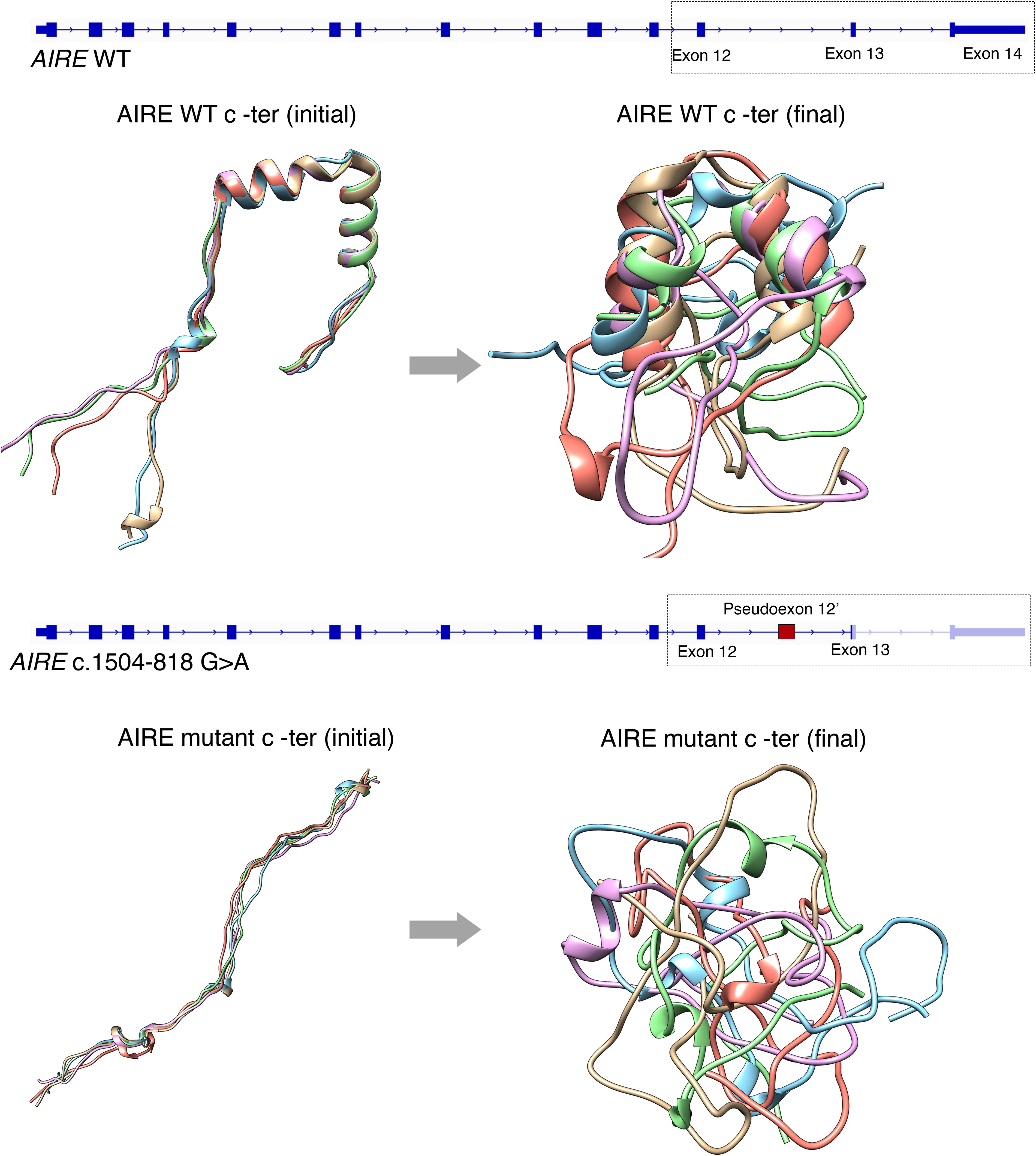
Protein modeling of the C-terminal region of AIRE. The starting and final conformations after dynamic protein simulations of C-terminus AIRE are shown. The top panel illustrates the structure of the 14 exons of wild-type (WT) AIRE and in silico modeling of the five most likely starting conformations of WT AIRE C-terminus region, each displaying two alpha helices. After 1 ms of dynamic protein simulations, the final conformations of these models are shown, retaining their alpha-helical structures at different orientations. The bottom panel presents mutant AIRE c.1504-818 G>A, resulting in an out-of-frame pseudoexon 12′ and an early termination codon in exon 13. The mutant AIRE models reveal the loss of discernible secondary structures in both starting and final conformations, indicating a disruption in proper protein folding due to the inclusion of the pseudoexon.
APECED is not amenable to hematopoietic stem cell transplantation and, although significant progress has been recently made with developing personalized immunomodulatory therapies targeting excess interferon-gamma responses (Oikonomou et al., 2024), there are currently no curative therapies for affected patients. Thus, we explored the use of antisense oligonucleotides (ASOs)—short, synthetic nucleotides that modulate mRNA splicing in the context of this deep intronic AIRE variant (Kim et al., 2023). ASOs bind to complementary sequences on pre-mRNA to block spliceosome interactions, promoting exon skipping. After testing several ASOs targeting the pathogenic pseudoexon, we identified one that completely restored wild-type AIRE transcript in vitro, marking the first genotype-specific therapeutic candidate for this disorder. We are in the process of testing the ASO in a knock-in mouse model with this AIRE variant, with the goal of evaluating its role as a genotype-specific therapy that may restore AIRE function in vivo.
What implications might this discovery in APECED patients have for the diagnosis and treatment of other monogenic diseases? GS data from a cohort of over 700 patients with childhood-onset, severe disease phenotypes revealed that deep intronic variants account for ∼7% of all molecular diagnoses (Wojcik et al., 2023). These variants, which cause disease through pseudoexon inclusion, have been sporadically reported in a wide range of diseases, including neurodevelopmental disorders, metabolic diseases, congenital heart disease, malignancy, benign hematological conditions, mitochondrial diseases, and other inborn errors of immunity (Vaz-Drago et al., 2017). However, systematic methods for detecting, prioritizing, and validating these variants are still largely underdeveloped. Furthermore, variant databases such as ClinVar are riddled with misclassified variants (i.e., false positives and negatives), and experimental evidence supporting variant pathogenicity is often lacking or based on poor quality evidence (Shah et al., 2018). As a result, the full extent of disease-causing deep intronic variants remains largely unknown, and what we and others have uncovered thus far may represent only the tip of the iceberg.
Our study underscores several key aspects of intronic variant detection, validation, and candidate therapy development. At the discovery stage, efforts should include precise phenotypic characterization, unbiased genetic sequencing, and orthogonal methods for variant identification and prioritization—beyond population frequency-based filtering—such as haplotype and AOH analyses, family segregation, and splice prediction tools (Fig. 2A). Variant validation is best achieved by demonstrating abnormal splicing in primary patient cells whenever possible, as biologically relevant splicing patterns are not always replicated in immortalized cell lines or through exon trapping (Wagner et al., 2023) (Fig. 2B). Functional evaluation is best achieved through assays that directly evaluate the gene’s function in relevant cell types (Fig. 2B). Validation efforts are critical to provide sufficient evidence for classifying variants as likely pathogenic, facilitating integration into genetic testing services such as gene panels, which are often used as first-tier diagnostic tools. Lastly, deep intronic variants offer a unique opportunity for ASO development, especially for diseases that are not corrected through hematopoietic stem cell transplantation or gene therapy (Fig. 2C).
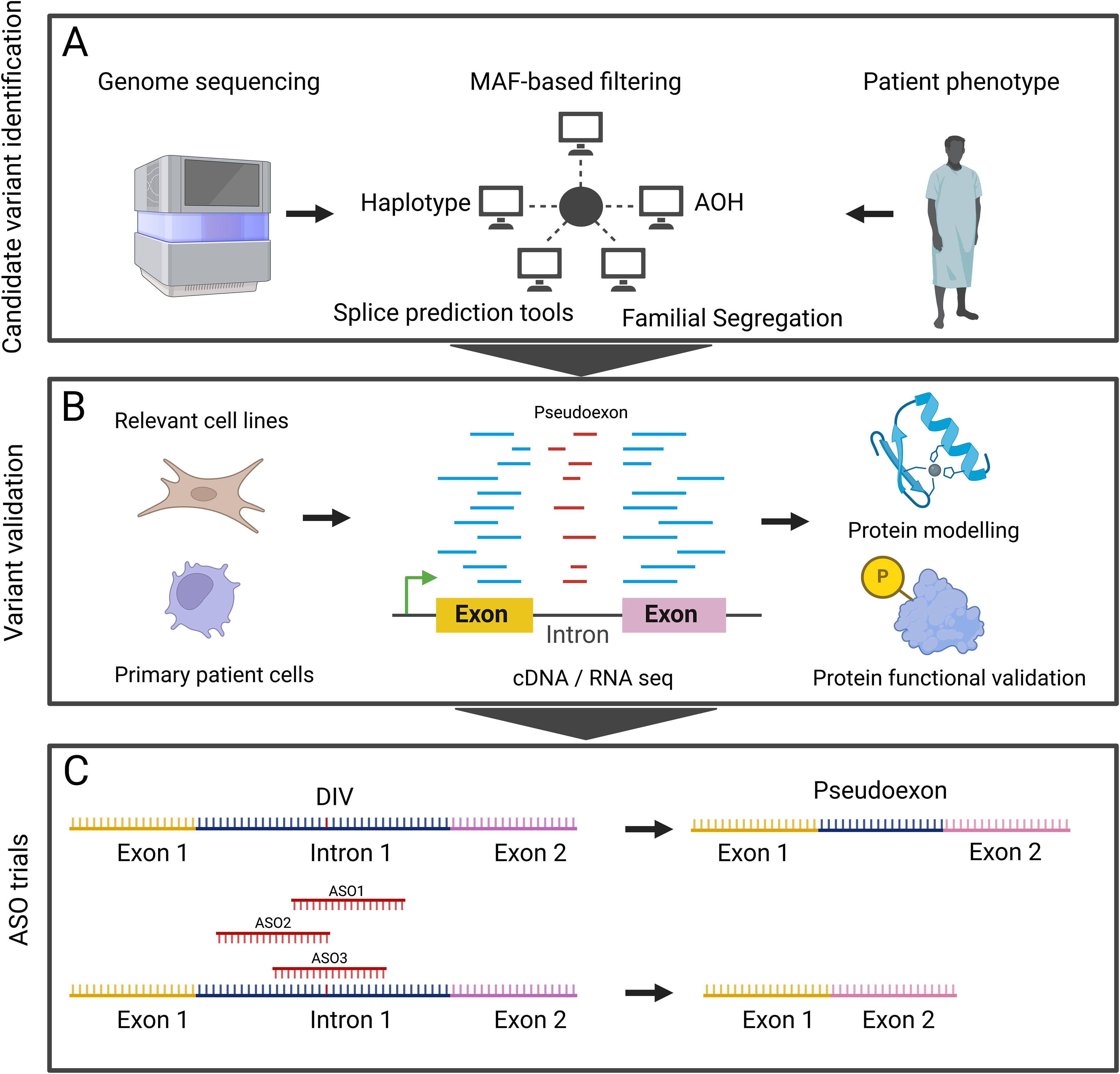
Process for intronic variant identification, validation, and antisense oligonucleotide development. The elements involved in the identification and validation of intronic variants, and generation and testing of candidate ASOs are shown. AOH, absence of heterozygosity; ASO, antisense oligonucleotide; MAF, minor allele frequency.
Splicing modulation via ASOs offers distinct advantages, including delivery to a broad range of cell types, no risk of insertional mutagenesis, and the elimination of the need for conditioning chemotherapy. A total of 15 ASOs have been authorized in different countries, for the treatment of spinal muscular atrophy, Duchenne muscular dystrophy, homozygous familial hypercholesterolemia, and primary hyperoxaluria type 1. In most cases, ASOs exert their therapeutic effects by inducing exon skipping to restore protein function, broadly reaching muscle cells, neurons, or liver cells (Lauffer et al., 2024). Phase III clinical trials are ongoing to evaluate ASOs for various monogenic diseases, including Angelman syndrome, Huntington’s disease, hemophilia, and monogenic amyloidosis (Lauffer et al., 2024).
While these examples highlight the potential of ASO therapy for addressing a range of genetic disorders, fully realizing the potential of ASO therapy will require pipelines that integrate deep intronic variant detection with scalable methods for variant validation, crucial for identifying novel pathogenic noncoding variants as therapeutic targets. Since many pathogenic deep intronic variants are private, financial incentives for development are lacking from the pharmaceutical industry. Moreover, ASO development requires significant trial and error, thus generating reliable bioinformatic tools to predict ASO-reversible sequences along with frameworks to enable the rapid development of personalized therapeutics from the bench to the bedside is essential (Fig. 2A–C). By accelerating ASO development, we can provide precision therapies for diseases like APECED, offering hope to patients with monogenic disorders currently lacking curative treatments.
Footnotes
Acknowledgment
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was funded by the Division of Intramural Research of NIAID/NIH.
Supplementary Material
Supplementary Data S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.