
Abstract
Bromodomain-containing protein 4 (BRD4) is a pivotal transcriptional regulator implicated in cancer, fibrosis, and inflammation, yet its phospho-regulatory network remains underexplored. This study leverages an extensive analysis of 1000 qualitative and 225 quantitative global phosphoproteome datasets to decode the BRD4 phosphorylation landscape. We identified S601 and S1117 as predominant phosphorylation sites, driving the majority of BRD4 phospho-signaling. Co-regulation analysis revealed 755 and 972 proteins positively cophosphorylated with S601 and S1117, respectively, including key interactors like TRIM28 (S473) and PRKAR2A (S78), which enhance transcriptional activity and cAMP signaling. Upstream kinases MAPK14 and GRK5 emerged as high-confidence regulators of S1117 and S601, respectively, with correlations in breast cancer highlighting disease relevance. In addition, 93 phosphosites in 71 transcription factors co-regulated with S1117 and 69 in 53 with S601 underscore the role of BRD4 in transcription control. These findings unveil a complex phospho-signaling network, offering novel therapeutic targets for BRD4-associated diseases and a foundation for future experimental validation.
Introduction
Bromodomain-containing protein 4 (BRD4) is a significant member of the bromodomain and extraterminal (BET) protein family. The BET family of proteins is primarily reported to be involved in various functions, including gene expression and cell cycle progression. Apart from BRD4, the other reported members of the BET family include bromodomain-containing protein 2 (BRD2), bromodomain-containing protein 3 (BRD3), and bromodomain testis-specific protein (BRDT) (Wang et al., 2021b).
Unlike BRDT, which is mainly found in the male germline, other proteins (BRD2, 3, and 4) are found to be expressed in different organs (Wei et al., 2024). BRD4 is reported in the literature as a 1362 amino acid long protein comprising an extra terminal domain and two bromodomains—BD1 and BD2. These domains assist in binding acetylated lysine residues on target proteins and histones (Drumond-Bock and Bieniasz, 2021; Wei et al., 2024). Each mentioned bromodomain comprises ∼110 amino acids in four alpha helices connected through loops (Travers et al., 2022). Apart from this, BRD4 also exhibits a serine-rich C-terminal phosphorylation site (CPS) and a C-terminal domain (CTD) known as the P-TEFb interaction domain (PID) (Itzen et al., 2014). The CTD region is predominantly needed for the interaction and recruitment of cyclin-dependent kinase 9 (CDK9) and CCNT1 subunits of the P-TEFb complex, decisive for transcriptional regulation (Jang et al., 2005; Itzen et al., 2014).
Biologically, BRD4 is associated with a myriad of functions like transcriptional co-regulation, epigenetic modification, and cellular process. As a transcriptional co-regulator, it aids in recruiting transcription factors (TFs) along with P-TEFb binding. Latter interaction facilitates the phosphorylation of RNA polymerase II (RNAPII), thereby promoting transcription initiation and elongation (Jang et al., 2005). The role of BRD4 as epigenetic regulators is majorly through the BD1 and BD2 bromodomains, which enable the protein to associate with active chromatin regions like enhancers and promoters. This association recruits acetyltransferases such as p300/CBP, which further acetylate histones, reinforcing an active chromatin state (Lasgorceix et al., 2014). In addition, a histone acetyltransferase activity of BRD4, which contributes to nucleosomal clearance and chromatin structure maintenance, was also reported. (Jang et al., 2005; Devaiah, Gegonne and Singer, 2016).
Besides the significant contributory roles in transcription and epigenetic regulation, BRD4 is also involved in enhancer-mediated gene expression. This is achieved through BRD4-TF interaction located at enhancers controlling the selectivity for specific target genes. Binding simultaneously to acetylated histones and TFs, BRD4 stabilizes TF occupancy at enhancers, enhancing transcriptional activity at these regulatory elements (Lasgorceix et al., 2014). BRD4 regulates cell cycle proteins such as CDK1 and CDK9 (Lasgorceix et al., 2014; Hu et al., 2022) and its inhibition leads to the prevalence of apoptosis and growth suppression highlighting the role of BRD4 in cell cycle regulation (Hu et al., 2022). Evidence on the role of BRD4 in inflammatory pathways like NF-κB signaling is also increasingly available (Jiang et al., 2017; Hu et al., 2022).
For modulating the above-mentioned functions of BRD4, phosphorylation plays an important part. For instance, BRD4 phosphorylation at the serine-rich CPS site enhances its interaction with the P-TEFb complex, including CDK9 and CCNT1, leading to transcription initiation and elongation (Drumond-Bock and Bieniasz, 2021; Lan et al., 2012). Similarly, phosphorylated BRD4 exhibits increased affinity for acetylated histones and DNA, mediated by its bromodomains. This enhances its ability to bind chromatin and associate with TFs like p53, thereby modulating gene expression (Lan et al., 2012; Drumond-Bock and Bieniasz, 2021). In diseases such as cervical cancer, hyperactive BRD4 phosphorylation supports uncontrolled growth (Rahman et al., 2011), and disrupting phosphorylation through inhibitors can halt proliferation, highlighting its regulatory significance (Rahman et al., 2011; Hu et al., 2022). Likewise, in fibrotic conditions such as cardiac fibrosis, phosphorylation changes the interaction dynamics of BRD4 with the fibrosis-related genes (Devaiah, Gegonne and Singer, 2016).
The currently available phosphoproteomic studies have delved into many aspects of the BRD4 regulatory mechanism. The phosphorylation of BRD4 at Y97 and Y98 is reported to increase the chromatin binding, reducing its affinity for BET inhibitors (BETi) and thereby contributing to resistance against these drugs (Wang et al., 2021). Similarly, phosphorylation of BRD4 at S1117 by IκB kinase‐α regulates its capacity to associate/dissociate to the chromatin and subsequent functions including DNA damage response (DDR) (Pecharromán et al., 2023). Another study on the direct binding on peptidyl-prolyl cis-trans isomerase NIMA-interacting (PIN1) to the T204 site of BRD4 reports the enhancement in the stability via ubiquitination inhibition. In addition, the role of PIN1 in catalyzing the isomerization of proline 205 of BRD4 and subsequent conformational change induction is also reported in the same study. This conformational change enhances the interaction of BRD4 with CDK9 and later its transcriptional activity (Hu et al., 2017).
These phosphorylation events collectively emphasize the significant role of BRD4 in orchestrating transcriptional and epigenetic regulation, influencing cell cycle progression, and contributing to diseases such as cancer, fibrosis, and inflammation. While BRD4 phosphorylation at specific sites like Y97 and S1117 is known, its broader phospho-signaling network remains unexplored. We hypothesize that cophosphorylation networks involving BRD4 underpin its transcriptional and disease-associated functions. Here, in this study, the cophosphorylation network of BRD4 is curated from the available literature, compiled, and cataloged to identify the upstream kinases and other binary interactors of BRD4 linking its activity to transcriptional regulation. This information provides a treasure trove of mechanistic, therapeutic, and systems-level insights on BRD4, which can be exploited by the scientific community for further validation and translational research.
Methods
Global phosphoproteomics dataset screening and BRD4 phosphopeptide assembly
To comprehensively investigate the phosphorylation landscape of BRD4, we conducted an extensive literature review by querying PubMed with the search terms “phosphoproteomics” OR “phosphoproteome,” applying stringent filters to exclude studies involving plants or review articles. Our focus was narrowed to high-quality, mass spectrometry-derived phosphoproteomics datasets originating from human cell lines, ensuring relevance to human cellular biology. Through meticulous manual curation, we identified global phosphoproteome datasets that reported Class-1 phosphosites, defined as phosphorylation sites with a localization probability of ≥75% and an A-score >13, reflecting high confidence in their identification and localization.
Our curation efforts specifically targeted datasets containing BRD4 phosphopeptides, which were generated through enzymatic digestion using LysC and/or trypsin, standard proteases in phosphoproteomics workflows. To systematically organize the datasets, we classified them into two distinct categories based on their experimental design: (1) profile datasets, which treated test and control conditions as independent entities, capturing all identified phosphopeptides in a comprehensive phosphosite profile and (2) differential profile datasets, which quantitatively compared phosphosite abundance between test conditions and their corresponding controls, highlighting differentially regulated phosphopeptides. This dual classification enabled a nuanced analysis of BRD4 phosphorylation dynamics under varying conditions.
For datasets in the differential profile category, we further analyzed Class-1 phosphosites to identify significant regulatory changes. Phosphosites were designated as upregulated if they exhibited a fold change of ≥1.3 or downregulated if they showed a fold change of ≤0.76, with statistical significance determined by study-specific parameters (p value <0.05). These thresholds ensured robust identification of biologically relevant phosphorylation events.
To ensure precise and standardized annotation of proteins and phosphosites across the curated phosphoproteomics datasets, we developed and implemented a robust, in-house mapping methodology tailored specifically for this study.
For protein-level mapping, each protein identified in the datasets was systematically linked to its corresponding gene symbol using the HUGO Gene Nomenclature Committee (HGNC) database, which provides a standardized and authoritative nomenclature for human genes. We utilized the most recent HGNC dataset available at the time of analysis to ensure that gene symbols were up-to-date and accurately reflected current nomenclature standards. This step was critical for harmonizing protein identifiers across diverse datasets, which often use varying naming conventions, and for facilitating downstream functional and comparative analyses.
At the phosphosite level, we mapped each identified phosphosite to its corresponding UniProt accession using the UniProt database release downloaded on May 13, 2023, as documented by the UniProt Consortium (2023). To achieve this, we employed a custom-built, in-house mapping tool. This tool was designed to parse phosphoproteomics datasets, extract phosphosite information (including amino acid position and sequence context), and align it with the appropriate UniProt protein sequence. The mapping process extracted phosphosite data from each dataset, including residue type (e.g., serine, threonine, and tyrosine), position within the protein sequence, and any associated metadata. Each phosphosite was assigned to the correct UniProt accession, accounting for protein isoforms. Validation checks flagged and resolved ambiguities, ensuring mapping accuracy. This approach provided a consistent and reproducible framework for BRD4 phosphosite analysis. To enhance the utility of the datasets for downstream analyses, we annotated each dataset with standardized metadata tags describing the biological and experimental conditions, following a structured format to facilitate efficient categorization and interpretation, as outlined in Sanjeev et al. (2024). This multifaceted approach—combining targeted literature curation, precise dataset classification, robust statistical analysis, and standardized mapping—enabled a comprehensive characterization of BRD4 phosphosites, laying a solid foundation for understanding their functional roles in human cellular processes.
Identification of predominant phosphosites representing BRD4 in cellular phosphoproteomes
The Class-1 protein phosphosites were derived from human cellular phosphoproteome profile datasets. The number of qualitative profile datasets where each protein phosphosite was observed or reported and the number of quantitative profile datasets where a protein phosphosite was discovered to be differently regulated were computed and ranked. Phosphosites found in over 50% of qualitative and quantitative profile datasets were considered as predominant phosphosites of BRD4. Phosphosites detected using phospho-antibodies or mutation-based methodologies were not identified or reported as Class-1 sites in these phosphoproteomics datasets and are excluded from this analysis.
Identification of protein phosphosites co-differentially regulated with major BRD4 sites
To identify the protein phosphosites/phosphosites in other proteins (PsOPS) that are positively and negatively co-differentially regulated with the predominant sites S1117 and S601 of BRD4, the differential regulation datasets corresponding to each phosphorylation site were categorized independently. Given the large number of datasets that contain various experimental conditions, biological systems studied, and the distinct analysis platforms employed, reanalysis of the raw datasets is impossible. Hence, for further analysis, the phosphosites were restricted to those with a localization probability of ≥75% and an ambiguity score (A-score >13). The following classifications were applied to each of these datasets. PsOPS (denoted as “o”) that were downregulated and upregulated with BRD4 (denoted as “b”) upregulation were designated as DoUb and UoUb. In contrast, those upregulated and downregulated with BRD4 downregulation were designated UoDb and DoDb. The PsOPs in UoUb and DoDb were considered positively co-regulated (in the same direction). In contrast, those in DoUb and UoDb were considered negatively co-regulated (opposite direction) with the expression of BRD4 phosphosite. These were also referred to as positively or negatively cophosphorylated PsOPs of the BRD4_S1117 and S601. Furthermore, Fisher’s exact test (FET) was carried out. We sorted the data based on the criteria such as FET p value <0.05, then a ≥10% ratio of Σ(nUbUo + nDbDo)/Σ(nUbDo + nDbUo) for positively co-regulated PsOPs and the ratio of Σ(nUbDo + nDbUo)/Σ(nUbUo + nDbDo) for negatively co-regulated phosphosites; a minimum of two distinct experimental conditions (experimental code count) that support positive or negative regulation; and a minimum of two distinct articles (PubMed IDs) that support positive or negative regulation (Priyanka et al., 2024; Lubaba et al., 2025; Mahin et al., 2025). The PsOPs filtered using the above-mentioned criteria were considered high-confidence proteins and used for subsequent analysis.
Assembly of upstream kinases and binary interactors
Among the filtered data, the upstream kinases of BRD4 were identified by the protocol of Johnson et al. (Johnson et al., 2023) through synthetic peptide screening to evaluate substrate specificity across the kinome with a 90th percentile cutoff and included in the analysis. The experimentally validated kinases and other phosphosite-specific interactions were retrieved by integrating data from multiple databases, including PhosphoSitePlus (Hornbeck et al., 2015), PhosphoELM 9.0 (Dinkel et al., 2011), RegPhos 2.0 (Huang et al., 2014), BioGRID (Oughtred et al., 2021), BIND (Bader, Betel and Hogue, 2003), HPRD (Keshava Prasad et al., 2009), and ConsensusPathDb release 35 (Kamburov and Herwig, 2022) (downloaded on May 22, 2023) (Sanjeev et al., 2024).
Retrieval of TFs associated with BRD4
To identify the TFs involved in the co-differential regulation of BRD4, we compared the list of TFs from the transcription factor database (TFDB) (Kanamori et al., 2004) with the proteins that are co-differentially regulated with the predominant sites S1117 and S601. The positively and negatively co-regulated proteins were extracted, and the TFs among these proteins were identified using the Venny tool available on Molbiotools. The upstream kinases of the identified TFs were predicted using in-house-pipelines (Johnson et al., 2023), NetworKIN (Linding et al., 2008), and AKID (Parca et al., 2019), and the union of these kinases was incorporated into BRD4 cophosphorylation networks, which were then captured for the further analysis.
Data visualization
For data visualization, lollipop plots were generated using the R/Bioconductor package trackViewer. The distribution of phosphosites across quantitative differential profile datasets was visualized using Python libraries such as Matplotlib and Pandas. Interaction networks were visualized in Cytoscape (version 3.10.) (Shannon et al., 2003), while dendrograms were created using RAWGraphs 2.0. The overall workflow of the study is summarized in Figure 1.
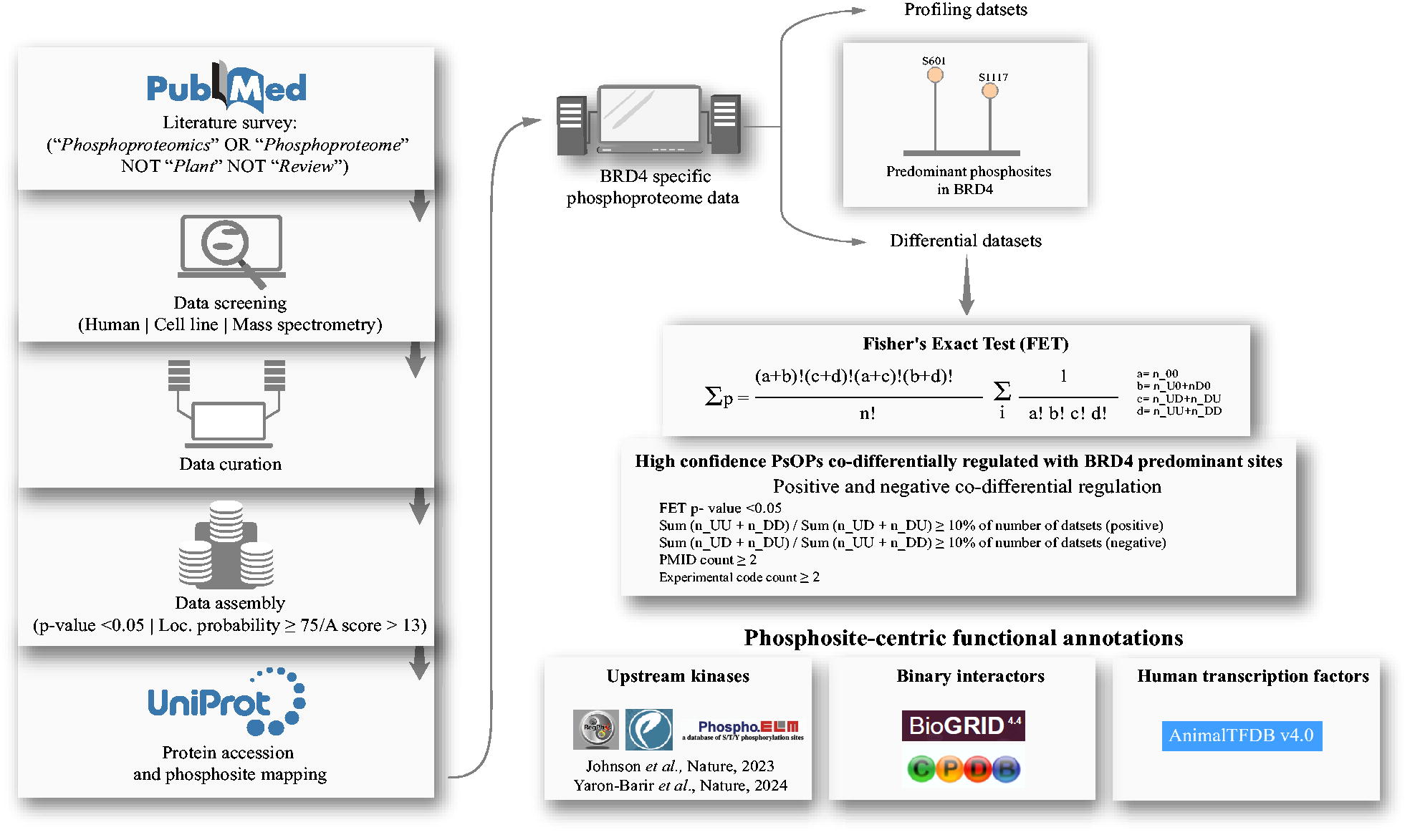
The methodology adopted in this study.
Results and Discussion
Analysis of BRD4 in global phosphoproteome datasets and identification of its predominant phosphosites
Over 3825 published human global cellular phosphoproteome datasets were screened, from which we sourced 1000 qualitative profiles and 225 quantitative differential datasets, which consist of Class-1 phosphosites represented on BRD4 phosphopeptides. The complete list of Class-1 BRD4 phosphosites in profile and differential datasets is given in Supplementary Table S1A, B. We identified 40 phosphosites from qualitative profiling datasets containing all the identified phosphopeptides and 29 from quantitative profiles determined to be differentially regulated. The phosphosites identified in around 50% of the datasets were regarded as predominant. Consequently, the phosphosites S601 and S1117 were selected as the predominant phosphosites based on their frequency of detection in 621 and 657 qualitative profiles and 102 and 54 quantitative differential datasets, respectively. The Class-1 BRD4 phosphosites detected in human cellular profile and differential datasets are represented in Figure 2.
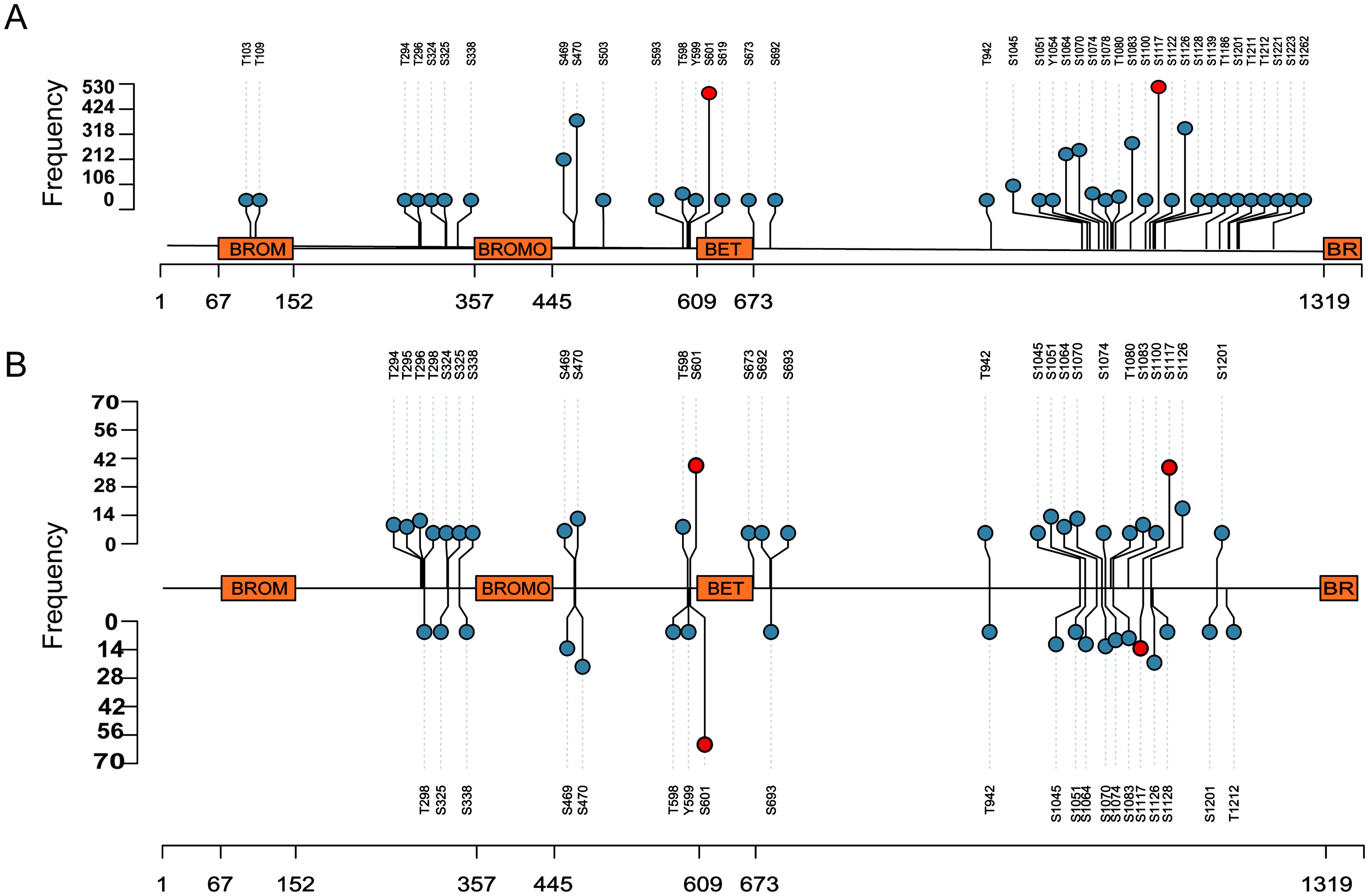
Lollipop plot with the BRD4 phosphosites in human cellular profiling and differential phosphoproteomics datasets.
Mutations such as E1113K, R1121H, and K1114E were identified in the phospho-motif regions of BRD4_S1117 using the ActiveDriverDB web tool (Krassowski et al., 2021) and are associated with various cancers, including bladder urothelial cancer, colon adenocarcinoma, and thymoma. Similarly, mutations like T598M and D605V occur within the phospho-motif region of BRD4 at S601 and have been linked to stomach adenocarcinoma and skin cutaneous melanoma. These mutations can disrupt phosphorylation processes, potentially contributing to disease progression. Phosphorylation of BRD4 at S1117 and S601 thus appears to be significant for its function and essential for BRD4-mediated cellular processes. Additional analysis was conducted further to understand the predominant phosphosites and their cophosphorylated proteins, and the findings are detailed in the following sections.
The identification of S601 and S1117 as predominant BRD4 phosphosites, supported by their high detection frequency across datasets, suggests their central role in phospho-signaling. Structurally, S601 lies near the BD1 domain, potentially modulating acetylated histone binding, while S1117, proximal to the PID, may enhance P-TEFb recruitment (Itzen et al., 2014; Pecharromán et al., 2023). The association of reported nearby mutations (e.g., E1113K) with cancer underscores their functional importance, possibly by altering kinase accessibility or chromatin affinity. Future structural studies, like X-ray crystallography, could elucidate these effects.
Phosphorylation sites in other proteins that exhibit a high degree of cophosphorylation with BRD4 sites, S601 and S1117
We analyzed the high-confidence PsOPs that are positively and negatively co-differentially regulated with the predominant phosphosites of BRD4 by applying a set of criteria such as FET p value <0.05, the ratio ≥10% of the number of positive co-expression/number of negative co-expression [n(UbUoDbDo/UbDoDbUo)] or the number of negative co-expression/number of positive co-expression [n(UbDoDbUo)/UbUoDbDo)], a minimum of two distinct experimental conditions (experimental code count), and a minimum of two distinct articles (PubMed IDs) that support positive and negative regulation.
Thus, we identified 755 and 26 PsOPs that are positively and negatively co-regulated with BRD4_S601, and 972 and 237 positively and negatively co-regulated PsOPs with BRD4_S1117 Supplementary Table S1C–F. The top PsOPs that were positively and negatively co-regulated with the predominant sites of BRD4 are as follows. Protein kinase cAMP-dependent type II regulatory subunit alpha (PRKAR2A) (S78 and S80) and serine/arginine repetitive matrix protein 2 (SRRM2) (T866) (positive co-differential regulation with BRD4_S1117) and PC4 and SFRS1 interacting protein 1 (PSIP1) (T141), insulin receptor substrate 2 (IRS2) (S1176), and echinoderm microtubule-associated protein-like 4 (EML4) (T899) (negative co-differential regulation with BRD4_S1117). Similarly, cyclin-dependent kinase 13 (CDK13) (S348), cyclin-G-associated kinase (GAK) (S829), and v-Abl Abelson murine leukemia viral oncogene homolog 2 (ABL2) (S631) are positively co-regulated with BRD4_S601, and SAM and domain-containing protein 1 (SASH1) (T101), pyridoxal-dependent decarboxylase domain-containing protein 1 (S779), and dynein cytoplasmic 1 light intermediate chain 1 (DYNC1LI1) (T515) are negatively co-regulated with BRD4_S601.
PRKAR2A encodes the protein kinase A (PKA) regulatory subunit IIα, which is essential for cAMP-dependent signaling and is among the constituents of the dormant state of PKA. By binding catalytic subunits, PRKAR2A keeps the enzyme dormant. The release of catalytic subunits by PRKAR2A upon cAMP binding permits the phosphorylation of downstream targets implicated in several cellular functions, including transcription, metabolism, and apoptosis (Sussman et al., 2020). The phosphosite S78 of PRKAR2A is reported to induce carcinogenesis in nonfunctioning pituitary tumors. Another cophosphorylated protein, SRRM2, is a nuclear-speckle marker that aids in the formation and maintenance of nuclear speckles, which are membraneless organelles involved in RNA processing (Xu et al., 2022). Although phosphosite-specific information is unavailable, TANK-binding kinase 1, which is increasingly linked to human cancers, has been reported as a regulatory protein of SRRM2 (Kim et al., 2013). RNA processing and DNA repair components are recruited to transcription sites by PSIP1, a chromatin protein linked to the transcriptional elongation complex exhibiting histone chaperone activity (Jayakumar et al., 2024). Another highly cophosphorylated protein was IRS2, which regulates signaling downstream of the insulin receptor and other receptor tyrosine kinases. The coordination of metabolic homeostasis depends on transduction via IRS2-dependent pathways, and many systemic insulin signaling abnormalities result from IRS2 dysregulation (Manohar et al., 2020). Hypo-phosphorylated EML4 is an essential protein for the formation and stability of microtubules, suggesting that the function of the protein is regulated by phosphorylation (Pollmann et al., 2006). Interestingly, another tumor suppressor protein observed among the PsOPs was SASH1, which is involved in apoptosis and cell division (Burgess et al., 2020). DYNC1LI1 plays a role in intracellular trafficking and chromosome segregation (Zhang et al., 2022). Another cophosphorylated protein, CDK13, plays a crucial role in transcription by phosphorylating the CTD of RNAPII, specifically at serine residues 2 and 5, which influences transcription elongation and RNA splicing (Pitolli et al., 2025). The phosphorylation of Ser2 in the heptad repeated by CDK9, CDK12, and CDK13 is necessary for release from transcriptional pausing, productive transcription, and cotranscriptional activities. These kinases play a part in cotranscriptional mRNA processing and transcription elongation (Quereda et al., 2019). In addition, GAK, a common serine/threonine kinase that helps vesicle traffickers uncoat clathrin (Lin et al., 2018), was found to be cophosphorylated with BRD4_S1117 at S829 phosphosite. v-Abl Abelson murine leukemia viral oncogene homolog 1 and ABL2, two ABL kinases, encourage tumor growth and metastasis in various solid malignancies. According to recent research, solid tumors exhibit elevated ABL kinase expression and/or activity, and metastasis is hampered by ABL inactivation (Greif, Burstein and Hammer, 1988).
Taken together, the extensive co-regulation of PsOPs with S601 and S1117 highlights the integration of BRD4 into diverse pathways. Positive correlations with TRIM28 (S473) and PRKAR2A (S78) suggest a synergistic role in DNA damage response and cAMP-mediated transcription, respectively (Agarwal et al., 2021; Sussman et al., 2020). Negative correlations with IRS2 (S1176) and SASH1 (T101) may indicate feedback mechanisms counteracting metabolic or tumor-suppressive signals. These networks, visualized using Cytoscape (Fig. 6), reveal important proteins like CDK13 and CDK7, which phosphorylate RNAPII CTD, reinforcing BRD4 transcriptional elongation role (Pitolli et al., 2025; Akoulitchev and Reinberg, 1998).
Upstream kinases predicted the predominant sites of BRD4 and their regulation in cancer
Although 51 phosphorylation sites are reported for BRD4 in the PhosphoSitePlus database (Hornbeck et al., 2015), protein kinases that regulate most of these phosphosites are currently unknown. JAK2 and CHUK are the currently known kinases that regulate BRD4, with JAK2 phosphorylating Y97 and Y98 and CHUCK phosphorylating S1117 (Pecharromán et al., 2023; Wang et al., 2021b). Phosphorylation at S1117 of BRD4 by CHUK is essential for the chromatin‐binding and DNA damage response activity of BRD4 (Pecharromán et al., 2023). Similarly, phosphorylation at Y97 and Y98 reduces BETi binding while enhancing the interaction with chromatin. In addition, the phosphorylation induces the interaction with STAT3 and promotes chromatin remodeling by concurrent binding with enhancers and super-enhancers to support a tumor-promoting environment (Wang et al., 2021).
However, phosphosites of these kinases were not identified to be consistently co-regulated with the predominant phosphosites in the current analysis. To further explore potential kinases that can regulate these predominant sites in BRD4, we utilized predicted kinases of BRD4 reported by Johnson et al. (2023) and predicted through kinase prediction tools. The current study identified two Johnson et al. predicted upstream kinases, mitogen-activated protein kinase 14 (MAPK14), which phosphorylates at Y182, T180, and S2, and G protein-coupled receptor kinase 5 (GRK5), which phosphorylates at T485 and S484 (Fig. 3; Supplementary Table S1C, F). MAPK14 was identified as the upstream kinase of BRD4_S1117, and the sites Y182, T180, and S2 were observed to be among the positively cophosphorylated PsOPs of BRD4 (S1117). The cophosphorylation of activity-inducing sites Y182 and T180 (Prickett and Brautigan, 2007) of MAPK14 along with BRD4_S1117 potentially highlights the phospho-signaling that regulates the transcriptional activity of BRD4.

The predicted upstream kinases of BRD4_S1117 and S601.
Similarly, GRK5 was predicted to phosphorylate BRD4 at S601. Its autophosphorylation sites S484 and T485 were found to be positively co-regulated with BRD4_S601 (Premont et al., 1994), further supporting the functional association between GRK5 and BRD4 phosphorylation. These findings provide strong evidence supporting the role of MAPK14 and GRK5 as potential upstream kinases that are capable of phosphorylating BRD4. In addition, phosphorylation of GRK5 at S484 and T485 strongly correlates with BRD4 phosphorylation at S601 in breast cancer, as reported by cProSite (Wang et al., 2023) (Fig. 4). This provides additional confidence to GRK5 as the potential upstream kinase of BRD4. Although serine/threonine-protein kinase B-raf (BRAF) was also predicted as a potential upstream kinase of BRD4 at S601, it was excluded from further analysis owing to the inhibitory role of its cophosphorylated site S365 (Cheung et al., 2008; Zhang and Guan, 2001; Guan et al., 2000).
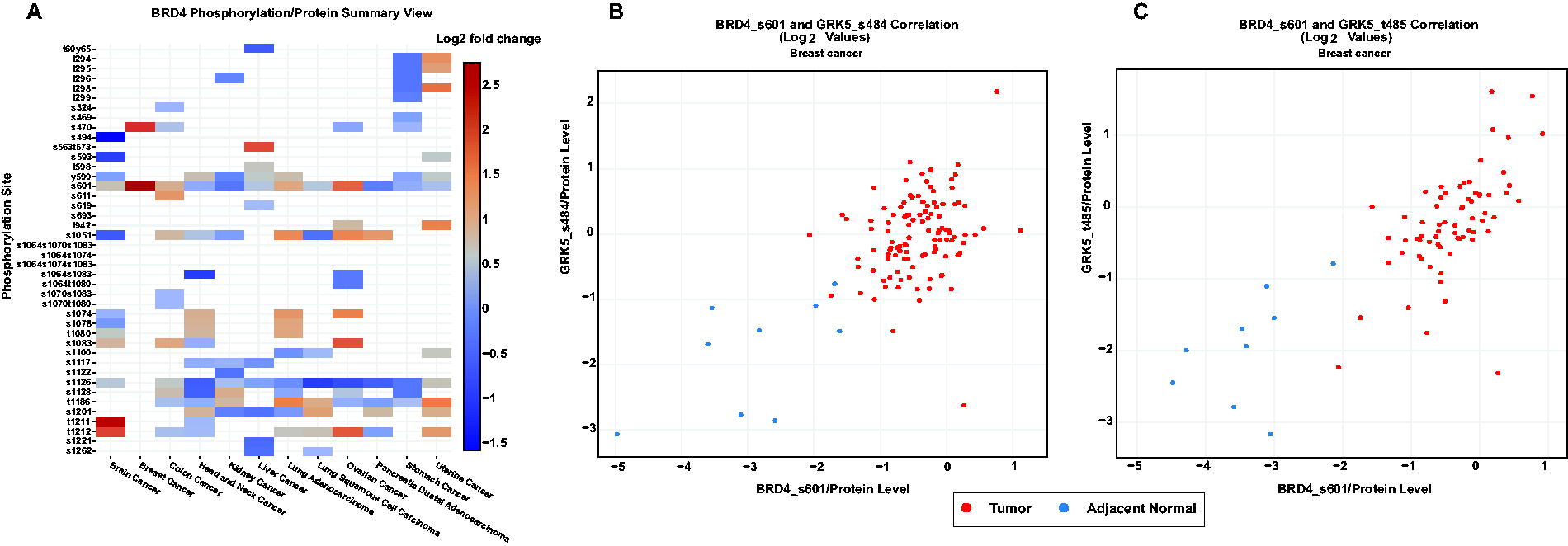
Tissue-level expression profiles and correlations of BRD4 and its phosphosites across multiple cancer types.
MAPK14, also known as p38α, is a member of the p38 MAPK family involved in cellular responses to stress and inflammatory cytokines. It plays a key role in regulating gene expression, cell differentiation, apoptosis, and autophagy (Madkour, Anbar and El-Gamal, 2021). Dual phosphorylation at Y182 and T180 is reported to be associated with the kinase activity of MAPK14 by Xu et al. (Xu et al., 2003). As the Y182 and T180 sites of MAPK14 are enzymatic inducing sites, the phosphorylation at these sites potentially induces the kinase activity of MAPK14 and regulates several physiological processes such as transcription, translation, and apoptosis. The physiological processes, such as transcriptional and translational control, cell cycle progression, cytoskeletal modeling, and cellular trafficking, are impacted by the phosphorylation of MAPK14 substrates (Brown and Wobst, 2022). Similarly, GRK5 is a serine/threonine kinase that belongs to the GRK family and is primarily involved in phosphorylating activated G protein-coupled receptors (GPCRs), leading to their desensitization and internalization. Beyond GPCR regulation, GRK5 also participates in various cellular processes, cardiovascular biology, inflammation and immunity, neurodegeneration, thrombosis, and hemostasis (Chaudhary and Kim, 2021). Notably, a study by Kunapuli et al. reported that lipids support the autophosphorylation and self-activation of GRK5 at the S484 and T485 sites (Kunapuli, Gurevich and Benovic, 1994). GRK5 moves into the cell nucleus, where it can interact with DNA and proteins unrelated to GPCRs to support “noncanonical” signaling, such as gene transcription (Marzano et al., 2021).
These findings provide strong evidence supporting the role of MAPK14 and GRK5 as potential upstream kinases that are capable of phosphorylating BRD4. Cophosphorylation of MAPK14 with S1117, via Y182/T180, aligns with its stress–response role, potentially amplifying BRD4 transcriptional output in inflammation or cancer (Madkour, Anbar and El-Gamal, 2021). Regulation of GRK5 on S601, supported by breast cancer correlations, suggests a novel link to GPCR signaling and tumor progression (Chaudhary and Kim, 2021). The exclusion of BRAF (S365) due to inhibition highlights the need for motif-specific validation. These findings propose MAPK14 and GRK5 as therapeutic targets, with inhibitors like SB203580 warranting preclinical testing in BRD4-driven cancers.
Phosphorylation in binary interactors associated with predominant sites of BRD4
The binary interactors associated with the predominant sites of BRD4 were analyzed by using HPRD (Keshava Prasad et al., 2009), BIND (Bader, Betel and Hogue, 2003), BioGRID (Oughtred et al., 2021), ConsensusPathDb release 35 (Kamburov and Herwig, 2022), CORUM (Tsitsiridis et al., 2023), and RegPhos 2.0 (Huang et al., 2014) databases. Thus, we identified 373 positively and 90 negatively co-differentially regulated binary interactors of BRD4_S1117. Similarly, 316 positively and 1 negatively co-differentially regulated binary interactor of BRD4_S601 were identified. The top 50 positively and negatively co-differentially regulated binary interactors of predominant sites of BRD4 are given in Figure 5. These interactions provide valuable insights into the complex regulatory networks associated with BRD4 phosphorylation sites, highlighting potential functional connections that may influence transcriptional dynamics and cellular processes.

The top 50 positively and negatively co-differentially regulated binary interactors of BRD4 phosphosites S1117 and S601.
Among the binary interactors, numerous transcription-associated proteins such as methyl CpG binding protein 2 (MECP2) (S80), nuclear receptor coactivator 3 (NCOA3) (S857 and S1330), negative elongation factor E (NELFE) (S51, S49, and S251), transcription intermediary factor 1-beta (TRIM28) (S473), apoptosis-antagonizing transcription factor (AATF) (S320 and S321), cyclic AMP-responsive element-binding protein 1 (CREB1) (S257), vimentin (VIM) (S339), heat shock protein beta-1 (HSPB1) (S78), and cyclin-dependent kinase 7 (CDK7) (S164) were positively cophosphorylated with BRD4_S1117. In addition, other proteins, including HSPB1 (S78), nuclear receptor corepressor 1 (NCOR1) (S2436), symplekin (SYMPK) (T1257), mediator of RNA polymerase II transcription subunit 1 (MED1) (T1032), heterogeneous nuclear ribonucleoprotein K (S284), RNA-binding motif protein 17 (RBM17) (T71), euchromatic histone lysine methyltransferase 2 (EHMT2) (T555), and PHD finger protein 8 (PHF8) (S120) were positively cophosphorylated with BRD4_S601. Notably, no transcription-associated proteins were identified among negatively co-differentially regulated interactors of either site.
MECP2 is a transcription regulator that binds to methylated DNA. Phosphorylation at S80 influences the interactions with chromatin factors such as HP1 and SMC3 and cofactors such as Sin3A and YB-1. These phospho-specific interactions contribute to the formation of MECP2-containing complexes capable of regulating gene expression (Gonzales et al., 2012). Similarly, S857 is a transcriptional activating phosphorylation site (Foulds et al., 2013). Under hypoxic stress, PFKFB4 phosphorylates S857 of NCOA3, and phosphorylation declines when PFKFB4 is reduced or absent. Lower phosphorylation of NCOA3 results in decreased activity in enhancing gene expression, thereby suppressing target gene activation (Dai et al., 2022). DYRK3 directly phosphorylates NCOA3 at S1330, disrupting its interaction with ATF4 and inhibiting ATF4 transcriptional activity, thereby preventing the activation of genes that promote purine synthesis (Ma et al., 2019).
Borisova et al. suggest NELFE (S49, S51, and S251) as a transcription-inducing site (Borisova et al., 2018). Similarly, the phosphorylation of TRIM28 at S473 induces its transcriptional activator activity, plays a crucial role in DNA repair, and promotes cell survival after DNA damage, serving as a molecular switch that controls gene expression (Agarwal et al., 2021). Phosphorylation of AATF at S320 and Ser321 enhances its interaction with histone H3, regulates histone acetylation, and subsequently influences gene expression and cell proliferation (Catena et al., 2021). VIM protein phosphorylation at S339 is associated with multiple signaling pathways, including PI3K/Akt, MAPK, Ras, JAK-STAT, and TGF-β. Downstream components of these pathways, such as AP-1, Stat3, NF-κB, and Smad2/3, act as potential transcriptional regulators of PD-L1. Phosphorylation of vimentin facilitates PD-L1 expression by recruiting these factors to PD-L1 promoter regions, thereby promoting immune escape and tumor survival (Jang et al., 2021). The function of HSPB1 is highly dependent on phosphorylation, which is primarily regulated at the transcriptional level. When conserved heat shock elements bind to preexisting heat shock TFs, phosphorylation at S78 becomes essential for nuclear translocation under stress, protecting nuclear structures and preventing apoptosis (Geum, Son and Kim, 2002). During mitosis, phosphorylation of CDK7 at S164 adversely affects TFIIH and reduces its transcriptional activity (Akoulitchev and Reinberg, 1998).
Casein kinase 2 (CK2) phosphorylates NCOR1 at S2436, stabilizing it against ubiquitin-dependent proteasomal degradation. The CK2-NCOR signaling network facilitates the invasive growth of human esophageal cancer cells by suppressing the transcription of interferon-γ–inducible protein 10 (Yoo et al., 2012). In the nucleus, SYMPK acts as a trans-activator to promote cell proliferation by regulating the transcription of multiple cell cycle-related genes through interactions with the nuclear factor YBX3. EGF signaling induces phosphorylation of SYMPK at T1257, followed by its nuclear translocation (Yoo et al., 2012; Zhang, Mao and Cao, 2017). During transcription, MED1 activates RNA polymerase II (Pol II) and dynamically associates with Pol II throughout transcribed genes, promoting Pol II recycling upon phosphorylation at T1032 by CDK9 (Chen et al., 2022). The phosphorylation sites RBM17_T71 (Al-Ayoubi et al., 2012; Chen et al., 2022), EHMT2_T555 (Dong et al., 2020), and PHF8_S120 (Arteaga et al., 2013) are also noteworthy. The identified binary interactors and their enriched biological processes are provided in Supplementary Table S1G–J. This analysis reveals numerous transcription-associated proteins cophosphorylated with BRD4 at S1117 and S601. These results provide critical new insights into BRD4-mediated regulatory networks that may influence gene expression.
To further elucidate the interaction network between the binary interactors of BRD4_S1117, we used the Signor 3.0 tool (Lo Surdo et al., 2023). Interestingly, among the binary interactome of BRD4_S1117, CDK2, PRKAA1, ATR, and EGFR exhibited multiple interactions with other proteins, indicating a regulatory role in the interactome network. A remarkable positive interaction flow was observed among CDK13, BRD4, POLR2A, CDK12, CDK7, CDK2, and FOXK2. Their numerous interactions indicate a complex regulatory network in which BRD4_S1117 may influence various cellular functions, including stress responses, transcription, and replication. The corresponding interaction network is depicted in Figure 6.
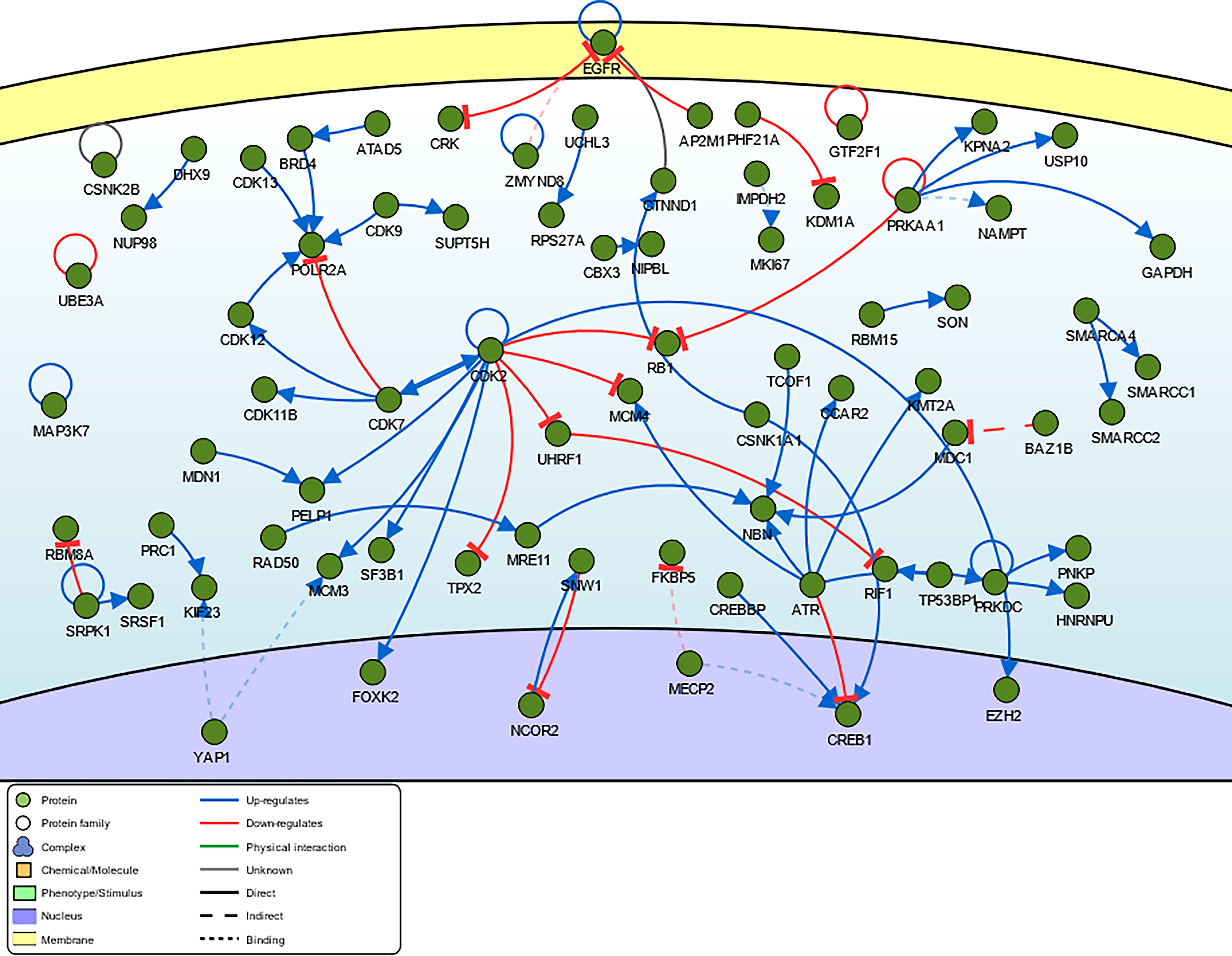
Interaction network of BRD4_S1117 and the cophosphorylated binary interactors. This figure illustrates the regulatory and functional relationships between BRD4_S1117 and binary interactors.
The predominance of transcription-related proteins among the binary interactome such as MECP2 (S80), NCOA3 (S857), FOXK2 (S373), NELFE (S51, S49, and S251), TRIM28 (S473), AATF (S320 and S321), NCOA3 (S1330), CREB1 (S257), VIM (S339), HSPB1 (S78), and CDK7 (S164) strongly suggests that BRD4 is deeply embedded in transcriptional regulation networks. These interactions and phosphorylation events likely play critical roles in modulating gene expression, chromatin dynamics, and cellular responses to external signals. By elucidating the phosphorylation-dependent interplay between BRD4 and its interactors, this study advances our understanding of the molecular mechanisms governing gene expression and cellular responses, offering new avenues for research and therapeutic intervention.
Many of the identified TFs were involved in processes that overlap with the functions of BRD4, including chromatin remodeling, transcriptional regulation, cell cycle control, and development. The phosphorylation of these TFs by kinases like MAPK14, CDKs, and others could modulate their interaction with BRD4 or their transcriptional activity, thereby influencing the regulatory networks in which BRD4 is a key player. This suggests a complex interplay between BRD4 and these TFs, mediated by phosphorylation events, which could be crucial for understanding the broader regulatory mechanisms.
Phosphosites in TFs cophosphorylated with major BRD4 sites
Identifying TFs cophosphorylated with BRD4 is strategic for elucidating the complex regulatory networks governing BRD4-associated gene expression. By mapping cophosphorylated TFs, we can uncover novel interactions and signaling crosstalk that influence the role of BRD4 in transcriptional regulation. Using the TFDB, our analysis identified 93 phosphosites in 71 TFs that were cophosphorylated with S1117, and 69 phosphosites in 53 TFs were phosphorylated with S601, establishing a transcriptional activity regulatory role for these phosphosites in BRD4 (Fig. 7, Supplementary Table S1K). A few resulting TFs are discussed below to provide insights into the TFs associated with BRD4.

The positively and negatively cophosphorylated transcriptional factors among the PsOPs of BRD4_S1117 and S601.The network is divided into three main sections: TFs cophosphorylated with BRD4_S601 (left), TFs phosphorylated with BRD4_S1177 (right), and TFs phosphorylated with both sites (center), represented by gray, yellow, and green nodes, respectively. Each TF is connected by edges to BRD4_S601 or S1177, with yellow nodes indicating positively cophosphorylated sites and green nodes indicating negatively cophosphorylated sites. TF, transcription factor.
AT-rich interactive domain-containing protein 1A (ARID1A) is the most often mutant chromatin regulator in all malignancies. ARID1A usually helps regulate active genes by controlling RNAPII, the enzyme that transcribes DNA into RNA. When ARID1A is lost, RNAPII struggles to pause properly, leading to disruptions in gene expression (Trizzino et al., 2018). It is crucial in inhibiting human-specific endogenous retrovirus H, whose aberrant activation can stimulate BRD4-dependent transcription and induce carcinogenesis (C. Yu et al., 2022a). Target gene expression is regulated by Forkhead box protein K1 (FOXK1), which supports several biological processes, such as the cell cycle, growth, proliferation, differentiation, programmed death, metabolism, DNA damage, drug resistance, angiogenesis, and carcinogenesis (M. Yu et al., 2022a). Nearly all clear cell renal cell carcinomas have genetically inactivated PBRM1, a subunit of the polybromo-associated BRG1-associated factor complex coactivator complex, which TFs employ to activate target genes (Gu et al., 2021). By suppressing p21cip1 transcription and hastening the cell cycle, motor neuron and pancreas homeobox 1 (MNX1) played an oncogenic role in cervical cancer (Zhu et al., 2020; Gu et al., 2021). The nuclear factor related to kappa-B-binding protein (NFRKB) is part of the metazoan INO80 complex, which is involved in chromatin remodeling. This process changes the shape of DNA packaging, which impacts transcription and gene accessibility (Peng et al., 2022). The TFs cophosphorylated with BRD4 predominant sites are essential for controlling chromatin remodeling, gene expression, and cell cycle progression. Their malfunction, due to mutation, aberrant activation, or altered phosphorylation, can cause transcriptional dysregulation and accelerate the development of cancer.
Further, we explored the upstream kinases of the TFs by using in-house pipelines such as Johnson et al. (Johnson et al., 2023), NetworkIN (Linding et al., 2008), and AKID (Parca et al., 2019). The top 50 predicted upstream kinases of the TFs were compared with the high confidence PsOPS that are positively and negatively co-regulated with the predominant sites of BRD4 (S601 and S1117) (Fig. 8). The TFs and their upstream kinases are provided in Supplementary Table S1L–N.

Predicted upstream kinases of the transcription factors identified among the positively and negatively cophosphorylated PsOPs of BRD4. PsOPs, phosphosites in other proteins.
Interestingly, among the positively and negatively co-regulated PsOPs of BRD4_S1117, few kinases were commonly observed, which included: MAPK14 (Y182), CDK13 (S383), CDK13 (T1246), CDK12 (S276), CDK7 (S164), DYRK1A (Y321), ATR (T1989), RPS6KA4 (T687) are positively and CSNK1A1 (T321) negatively co-regulated with BRD4_S1117. STK39 (S385) and ATR (T1989) are positively co-regulated with BRD4_S601. No upstream kinases were found to be negatively co-regulated with BRD4_S601.
MAPK14 (Y182), predicted to be the upstream kinase of BRD4_S1117, also regulates other TFs, such as ARID1A (S363) and FOXK1 (S472) among the PsOPs. Similarly, other TFs regulated by these kinases include PBRM1 (S1453, T9, S10, S948), TCF12 (S67), FOXK1 (S472), SP1 (S7), MNX1 (S77), and NFRKB (S228). The inhibition of CDK12 leads to the loss of key elongation factors from POL II transcription units, underscoring its essential role in maintaining transcriptional integrity. CDK12 also phosphorylates POL II CTD at Ser2 and Ser5 in human cells, emphasizing its crucial involvement in transcriptional control (Fan et al., 2020). Alongside CDK12 and CDK13, CDK7 functions as an effector kinase that phosphorylates POL II and other components of the transcriptional machinery. This activity is directly influenced by phosphorylation at S164, which modulates CDK7 function in transcription initiation. Moreover, CDK7 is intimately associated with the transcription initiation factor TFIIH, contributing to the regulation of transcription initiation (Düster et al., 2024; Fisher, 2019).
Another key player, DYRK1A, is preferentially recruited by RNAPII to the promoters of actively transcribed genes. It recognizes a conserved palindromic motif significantly enriched in DYRK1A-bound promoter regions and is necessary for DYRK1A-dependent transcriptional activation. DYRK1A phosphorylates the CTD of RNAPII at Ser2 and Ser5, further establishing its role as a transcriptional regulator and CTD kinase (Di Vona et al., 2015). ATR, phosphorylated at T1989, is also an activity-inducing site (Nam et al., 2011). It is a DNA damage-regulated kinase involved in the DDR pathway. Cellular responses to stress and DNA damage are coordinated by ATR, one of the two primary regulators of the DDR pathway. These responses include transcriptional regulation, DNA repair, activation of cell cycle checkpoints, and, if necessary, induction of apoptosis (Jackson and Bartek, 2009; Cimprich and Cortez, 2008; Sancar et al., 2004; Barnieh, Loadman and Falconer, 2021).
ATR (T1989) and STK39 (S385) were identified as the positively cophosphorylated upstream kinases of BRD4_S601. Phosphorylation of ATR at S1989 is critical for its activation (Hanaki, Habara and Shimada, 2021) and regulation of DNA damage (Nam et al., 2011). The DNA damage response is primarily regulated by the ATR and ATM protein kinases, which signal to regulate apoptosis, DNA replication, cell cycle transitions, and DNA repair (Cimprich and Cortez, 2008). Compared with adjacent tissues, clinical hepatocellular carcinoma (HCC) tissues exhibited significantly higher levels of STK39 expression, which was linked to a decreased chance of patient survival and was influenced by the transcription factor SP1. Along with ATR (T1989) and STK39 (S385), BRD4 is phosphorylated at S601, highlighting its involvement in the response to DNA damage and cancer progression.
Taken together, the CDK13, CDK12, CDK7, MAPK14, and DYRK1A, which are essential regulators of transcription, were found to be among the BRD4 cophosphorylated kinases underscoring the regulatory role of BRD4 in transcription and its potential in signaling pathways involving these TFs. Modulating POL II CTD phosphorylation and facilitating both transcription initiation and elongation plays vital roles in maintaining gene expression fidelity. Also, the phosphorylation of BRD4 at S601 and S1117, alongside the coordinated activation of kinases ATR (T1989) and STK39 (S385) implies a pivotal role in modulating the transcriptional regulatory activity like DNA damage response and cancer progression (e.g., HCC) Understanding their functional mechanisms provides valuable insights into the broader regulatory networks that govern transcriptional processes. The cophosphorylation of 71 TFs with S1117 and 53 with S601, including ARID1A and PBRM1, underscores the influence of BRD4 in chromatin remodeling, particularly in cancers with frequent mutations (Trizzino et al., 2018; Gu et al., 2021). Overlapping kinases (e.g., CDK12, DYRK1A) suggest a coordinated phospho-regulatory hub, critical for maintaining transcriptional fidelity (Fan et al., 2020; Di Vona et al., 2015). The disruption of these networks in diseases like HCC (via STK39) opens avenues for combinational therapies (e.g., BRD4i with kinase inhibitors), though in vivo validation is essential. This in silico study is hypothesized on the available phosphoproteome datasets and therefore limits causal inference, as co-regulation does not confirm direct phosphorylation. Future research should validate MAPK14/GRK5 interactions via kinase assays and assess the S601/S1117 mutants’ impact on chromatin binding using ChIP-seq. Single-cell phosphoproteomics could further resolve heterogeneity in cancer contexts.
Computational validation
Additional computational validation steps were performed to robustly validate our findings using existing data and in silico approaches, ensuring confidence in our results.
To benchmark the reliability of our identified BRD4 phosphosites, we cross-referenced our findings with the PhosphoSitePlus database, a widely recognized resource for phosphosite annotations. For example, the phosphosite S601 was reported in 40 HTP studies in PhosphoSitePlus, while our analysis detected it in 621 qualitative and 102 quantitative differential experimental conditions, indicating robust detection across diverse contexts. Similarly, S1117, documented in 1 LTP and 73 HTP studies in PhosphoSitePlus, was identified in 657 qualitative and 54 quantitative differential conditions in our datasets. Notably, our curation uncovered several novel BRD4 phosphosites not previously reported in PhosphoSitePlus, underscoring the comprehensiveness of our approach. A complete list of detected phosphosites, their frequencies across experimental conditions, and their HTP/LTP counts from PhosphoSitePlus is provided in Supplementary Table S2A.
To further validate our computational findings, we conducted a protein–protein interaction (PPI) analysis using the STRING database (Szklarczyk et al., 2019). The analysis of 780 BRD4 binary interactors revealed a robust and biologically significant interaction network, comprising 402 nodes and 1816 edges (Fig. 9), with a highly significant PPI enrichment p value (<1.0e-16). The experimental (604 edges) and text mining (572 edges) (Supplementary Table S2B) provided the most substantial evidence, underscoring the reliability of these interactions and their alignment with well-documented BRD4 functions in transcriptional regulation and chromatin remodeling. The high connectivity, reflected by an average node degree of 9, indicates that BRD4 interactors form densely interconnected functional modules, likely representing key regulatory complexes such as transcriptional machinery or epigenetic modification hubs. Notably, the significant enrichment of edges over the expected number (814) suggests that these interactions are not random and are likely driven by coordinated cophosphorylation events, reinforcing the functional relevance of the identified phosphosites. The predominance of experimental and database (323 edges) evidence further supports the potential for direct physical interactions, making these interactors prime candidates for experimental validation in BRD4-related pathways. In addition, the reduction from 780 input proteins to 402 nodes highlights the possibility of novel or less-characterized interactors, which could be explored to uncover new regulatory mechanisms. Network analysis of this dense network structure using the Cytoscape (Shannon et al., 2003) network analyzer module (Assenov et al., 2008) also revealed that BRD4 acts as a central hub (degree centrality, closeness centrality, and betweenness centrality) (Supplementary Table S2C), orchestrating multiprotein complexes critical for cellular processes like cell cycle progression and oncogenesis.

Protein–protein interaction network of the binary interactors of BRD4 identified among the positively and negatively cophosphorylated PsOPs.
Discussion
This study provides a comprehensive analysis of the phosphorylation landscape of BRD4, uncovering a complex cophosphorylation network that underscores its pivotal role in transcriptional regulation and disease pathogenesis. By curating 1000 qualitative and 225 quantitative phosphoproteome datasets, we identified S601 and S1117 as predominant phosphosites, detected in 621 and 657 qualitative profiles and 102 and 54 quantitative differential datasets, respectively. These sites, supported by their high detection frequencies and alignment with PhosphoSitePlus annotations (40 HTP studies for S601, 1 LTP, and 73 HTP for S1117), appear to be primal to phospho-signaling of BRD4. The discovery of 755 and 972 proteins cophosphorylated with S601 and S1117, respectively, including key interactors like TRIM28 (S473) and PRKAR2A (S78), and the identification of MAPK14 and GRK5 as upstream kinases, offer novel insights into the regulatory mechanisms of BRD4. Furthermore, the co-regulation of 93 phosphosites in 71 TFs with S1117 and 69 in 53 TFs with S601 highlights the influence of BRD4 on chromatin remodeling and transcription, particularly in cancer contexts.
The identification of S601 and S1117 as predominant phosphosites augments their structural and functional significance. S601, located near the BD1 domain, likely modulates the binding of BRD4 to acetylated histones, enhancing its recruitment to active chromatin regions (Wang et al., 2012). Conversely, S1117, proximal to the PID, may facilitate the recruitment of components critical for RNAPII phosphorylation and transcriptional elongation (Fujinaga, Huang, and Peterlin, 2023). These findings are consistent with previous reports, such as that of Pecharromán et al. (2023), which linked S1117 phosphorylation by IKK‐α to chromatin binding and DDR and underscore the role of sites in BRD4-mediated cellular processes. The association of nearby mutations (e.g., E1113K and T598M) with cancers like bladder urothelial carcinoma and stomach adenocarcinoma further suggests that disruptions in these phospho-motifs may contribute to oncogenesis by altering kinase accessibility or chromatin affinity.
The comprehensive cophosphorylation networks identified in this study reveal the integration of BRD4 into diverse cellular pathways. Positive co-regulation with TRIM28 and PRKAR2A suggests synergistic roles in DDR (Li et al., 2024) and cAMP-mediated transcription (Oyen et al., 1989), respectively. TRIM28, a transcriptional regulator, enhances DNA repair and chromatin remodeling, probably enhancing the epigenetic functions of BRD4, while the involvement of PRKAR2A in cAMP signaling may link BRD4 phosphorylation to metabolic and apoptotic pathways. Negative co-regulation with proteins like IRS2 (S1176) and SASH1 (T101) indicates possible feedback mechanisms that counteract metabolic (Santamaria et al., 2015) or tumor-suppressive signals (Burgess et al., 2020), highlighting the dynamic balance in the regulatory network of BRD4. The cophosphorylation of transcription-associated proteins, such as CDK13 (S348), CDK7 (S164), and MECP2 (S80), with S601 and S1117 reinforces the role of BRD4 in transcriptional elongation and chromatin organization, as these proteins collectively modulate RNAPII activity and gene expression (Even et al., 2016; Chou et al., 2020; Song et al., 2014).
To further validate these cophosphorylation networks, we performed PPI and signaling pathway analyses using the STRING database, focusing on high-confidence binary interactors that were cophosphorylated. The STRING analysis revealed a significantly enriched PPI network (p value <1.0e-16) involving BRD4 and interactors like CDK7, CDK13, and TRIM28, centered on transcriptional regulation and chromatin remodeling.
The prediction of MAPK14 and GRK5 as upstream kinases for S1117 and S601, respectively, introduces novel regulatory mechanisms for BRD4. MAPK14, a stress-responsive kinase, likely amplifies transcriptional activity of BRD4 in inflammatory or oncogenic contexts through cophosphorylation at Y182 and T180. In oncogenic contexts, BRD4 drives the expression of oncogenes like MYC, and MAPK14 contributes to cancer cell proliferation and survival (Wang et al., 2017). Its role in phosphorylating TFs like ARID1A (Wang et al., 2017; Rehman et al., 2022) further suggests a coordinated phospho-regulatory hub that integrates BRD4 with wider transcriptional networks. GRK5, traditionally associated with GPCR desensitization, may regulate chromatin interactions of BRD4 via S601, correlating with BRD4 phosphorylation in breast cancer. This finding, supported by cProSite correlations, opens new avenues for exploring GPCR signaling in BRD4-driven cancers (Wu et al., 2019).
The co-regulation of 71 TFs with S1117 and 53 with S601, including ARID1A, PBRM1, and SP1, underscores the role of BRD4 in chromatin remodeling and transcriptional control, particularly in cancers with frequent mutations in these TFs (Mandal et al., 2022; Brugarolas, 2013; Vizcaíno, Mansilla and Portugal, 2015). Overlapping kinases, such as CDK12, CDK13, and DYRK1A, suggest a tightly coordinated phospho-regulatory network that maintains transcription by phosphorylating RNAPII and associated factors (Fan et al., 2020; Di Vona et al., 2015). The involvement of ATR (T1989) and STK39 (S385) in S601 co-regulation further indicates BRD4 in DDR (Fokas et al., 2014) and cancer progression, particularly in breast cancer, where STK39 overexpression is linked to poor prognosis (Qiu et al., 2021). These findings propose BRD4 as a potential therapeutic target, with combinational strategies involving BRD4 inhibitors and kinase inhibitors warranting further investigation.
Despite the robustness of our findings, several limitations must be acknowledged. The study relies on computational analyses of existing phosphoproteome datasets, which, while comprehensive, limit inference. Cophosphorylation does not confirm direct phosphorylation or functional interactions, and the heterogeneity of experimental conditions across datasets may introduce variability. The absence of wet lab validation, due to resource constraints, is a notable limitation. We postulate this work as a foundation for future experimental validations to confirm the identified interactions and their functional consequences.
In conclusion, this study illuminates the phospho-regulatory network of BRD4, positioning S601 and S1117 as critical nodes in transcriptional and disease-associated pathways. The integration of cophosphorylated proteins, upstream kinases, and TFs into coherent networks, validated through STRING analyses, underscores the role of BRD4 as a master regulator. While computational limitations and the lack of wet lab validation necessitate cautious interpretation, the proposed future experiments and therapeutic strategies offer a roadmap for advancing BRD4 research. These findings not only deepen our understanding of molecular mechanisms of BRD4 but also pave the way for targeted interventions in cancer, fibrosis, and inflammation, heralding a new era of precision medicine.
Conclusion
This study unveils the intricate cophosphorylation network of BRD4, illuminating its role as a potential master regulator of transcription and a potential therapeutic target in diseases like cancer and fibrosis. By analyzing an expansive dataset of 1000 phosphoproteome profiles and 225 differential datasets, we pinpointed S601 and S1117 as pivotal phosphorylation sites, driving a complex web of interactions with 755 and 972 co-regulated proteins, respectively. The identification of MAPK14 and GRK5 as upstream kinases, validated by breast cancer correlations, offers novel entry points for precision medicine, while the engagement of 71 and 53 TFs with S1117 and S601, respectively, underscores the importance of chromatin remodeling competence of BRD4. These findings not only deepen our understanding of BRD4 phospho-signaling but also chart a transformative path toward targeted therapies, such as MAPK14 inhibitors, to disrupt aberrant transcriptional networks. Future experimental validation—through kinase assays, phospho-mutant models, and single-cell analyses—will be crucial to translate these insights into clinical breakthroughs, heralding a new era in BRD4-targeted interventions.
Footnotes
Acknowledgments
The authors acknowledge Yenepoya (Deemed to be University), Mangalore, for providing infrastructure for the Centre for Integrative Omics Data Science.
Authors’ Contributions
This study was designed by R.R. A.C.L. performed the analysis and drafted the article. A.C.R. and R.R. supervised the work. A.C.L. wrote the article with editing assistance from F.L. and A.C.R. P.B.S. assisted in data mapping and assembly. A.P.G., A.M., and S.S. assisted in data curation and figure generation. All the authors have read and approved the final article.
Availability of Data and Materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Author Disclosure Statement
The authors declare that they have no competing interests.
Funding Information
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.