
Abstract
Background:
Stress urinary incontinence (SUI) is a widespread and frustrating condition that affects millions of people worldwide, with severe consequences on patients’ quality of life and health care systems’ costs. Currently, the most severe cases of SUI are treated using implanted (and rather invasive) extraurethral artificial sphincters. The authors propose an innovative, minimally invasive endourethral device for the treatment of SUI.
Methods:
Ten patients with SUI were enrolled in three Italian centers and underwent device implantation. After 10, 30, 60, and 90 days, correct device position was confirmed by ultrasonography. Improvements in continence and quality of life were evaluated through a 24-hour pad-test, an International Consultation on Incontinence Questionnarie-Short Form (ICI-Q) and a custom checklist. The device was explanted after 90 days.
Results:
The proposed device was successfully implanted and explanted in 8 out of 10 patients. The results of the pad-test, ICI-Q, and custom checklist demonstrated remarkable improvements in continence (median improvement: 82% with respect to the initial condition) and quality of life (mean reduction of the impact of urine losses on the quality of life: 61%). No major pain or discomfort was reported.
Conclusions:
The results demonstrate the efficacy of the proposed endourethral artificial sphincter in addressing SUI. The proposed device was successfully implanted and explanted in a short time (∼10 minutes) without intrinsic side effects and without triggering pain or discomfort.
Introduction
Urinary incontinence (UI) is a severe problem affecting people’s quality of life. It often leads to social withdrawal and massive restrictions in various lifestyle habits (i.e., social, occupational, domestic, and psychophysical well-being). The incidence and prevalence of this pathology are often underestimated, as UI is still often considered a taboo. 1
Stress urinary incontinence (SUI), defined as the occurrence of involuntary urine leakages during bladder filling owing to failure in the urethral closure mechanism, affects 39.2% of patients with UI and leads to emotional and psychological stress. Global statistics report that approximately 167 million male and female patients in 2018 suffered from SUI. 2 As a consequence, this medical condition is a significant challenge for the health care systems, also in terms of financial resources. The risk factors for SUI differ between males and females: whereas pregnancy, vaginal delivery, and age are the main risk factors in women, SUI in men is less common and often associated with surgeries of the lower urinary tract. 3 Upon proper diagnosis, the treatment plan for SUI can include three stages: conservative, pharmacological, and surgical ones. 4 In most cases, patients are treated with a first-line regimen of drugs, possibly in combination with specific exercises and electrophysiological stimulation. When conservative options fail, minimally invasive surgery is applied. However, standard surgeries, especially the application of implants, do not pursue any causal therapy. Nonabsorbable meshes and ligaments have fallen into disrepute owing to complications. A structured and successful treatment of incontinent patients is made possible by numerous guidelines, 5 including the European Association for Urology ones, which are constantly updated. 6,7 Despite standardization and recommendations, nowadays, SUI is still not causally curable. 8
To approach severe SUI, extraurethral or endourethral artificial urinary sphincters (AUS) have been proposed. 9 –11 They are prostheses implanted around the urethra or within the urethral lumen, respectively, by surgical and endoscopic approaches, able to replace the functions of the biological sphincter. The gold standard of extraurethral AUS is the AMS 800 (Boston Scientific, Marlborough, MA, USA). It is an implantable fluid-filled device placed around the urethra that allows the patient to control urine voiding. 12 Although it represents the gold standard, it presents several issues including invasiveness, requirement of a relatively long surgical procedure, suitability only for men, side effects, and relatively high cost. 13
Few endourethral devices have been investigated for their ability to reduce the invasiveness of the implant procedure. Examples include the FemSoft system (Rochester Medical Corp, Stewartville, MN, USA) 14 and the InFlow system (Vesiflo, Redmond, WA, USA). 15 Such endourethral devices can be used only by female patients, they show urine compatibility issues, and in general are not designed for long-term use. Recently, some of the authors proposed an innovative magnetically controlled endourethral device. It was a unisex, minimally invasive, low-cost, and patient-compliant magnetic device consisting of a magnetic system and a polymeric valve. Details of the design process and previous experimental results are available in Mazzocchi et al. 16,17 The device has been already characterized through laboratory and bench tests, cadaver tests, and a 28-day first-in-human trial on six patients. 18 The results of such first pilot study demonstrated the feasibility of the approach and no significant pain or discomfort due to the implant. However, the device showed the tendency to migrate toward the bladder because of the lack of an anchoring system. The migration was probably caused by an oversized external magnet causing excessive forces that dislocated the device and by the anatomical variability of patients’ urethral diameter. In this study, we show a redesign of the device, which includes an anchoring stent to prevent migration. Furthermore, we show the results of a clinical evaluation of the device efficacy carried out in three different Italian centers, involving 10 patients. The duration of the observation period was 90 days, triple than the previous study. Results concern safety, acceptability, implantation efficacy, and efficacy of the device in restoring urinary continence.
Materials and Methods
To address the issues emerged in the previous study, 18 the authors optimized the design of the proposed device, as detailed in the Supplementary Data S1 and S2.
Once the final design was selected, a clinical evaluation was conducted to demonstrate the safety, acceptability, and efficacy of the proposed device in restoring urinary continence. A prospective multicentric trial was designed for this purpose. The selected centers were Campus Bio-Medico University of Rome (Rome, Italy, coordinator center), Casa di Cura San Camillo (Forte dei Marmi, LU, Italy), and Azienda Ospedaliero Universitaria Pisana (Pisa, Italy). From August 17, 2022 to August 16, 2023, 10 patients were enrolled in a pilot study. The device remained implanted for 90 days.
Encrustations still represent an open issue for several medical devices to be employed in the urinary system, as their permanence in a rather hostile environment, being in continuous contact with the urine, often leads to failure in a few weeks. The device proposed in this study remained implanted and fully functional for 90 days. This behavior is corroborated by previous evidence. 19 In such a study, the performance of bare polydimethylsiloxane (PDMS) unidirectional valves (identical to the ones used in this paper) was evaluated. The valves were tested both in terms of fatigue behavior and opening pressure over 126 days of immersion in different types of urine with daily openings. Results showed that PDMS-based valves did not vary their performance over time, in terms of opening pressure, and a few encrustations could be found on their surface. Some authors have claimed that sufficient resistance to encrustations for at least two months can be considered suitable for long-term application in the urinary tract. 20 Putting together the in vitro evidence shown in Mazzocchi et al. 19 over 126 days and the clinical evidence reported in this study over three months, we argue that the proposed device can remain in the urethra keeping its stability and functionality for a long period (at least 12 months, or even more).
The pilot study was approved by the Ethical Committee of Campus Biomedico of Rome (02/22 PAR ComEt CBM) on February 18, 2022, by the Ethical Committee of AREA VASTA NORD-OVEST for Casa di Cura San Camillo (21999_Cecchi) on April 07, 2022 and for Azienda Ospedaliero Universitaria Pisana (21999_Pomara) on May 19, 2022 and by the Italian Ministry of Health on June 15, 2022 (DGDMF/P/I.5.i.m.2/2022/2044). The primary objectives of the study were: (1) to assess the safety and reproducibility of the implantation procedure for the magnetic endourethral device; (2) the quantitative restoration of urinary continence; (3) whether the patients tolerated the procedure; (4) if possible, whether irritation or other symptoms resulting from the use of the device emerged.
The inclusion criteria for the trial were: (1) male patients aged 45–80 years diagnosed with SUI secondary to prostate surgery in whom medical and rehabilitative treatment had failed; (2) female patients aged 45–80 years diagnosed with SUI in whom medical, rehabilitative, and surgical treatment had failed; (3) sterile urine culture; (4) urodynamic test showing evidence of sphincter dysfunction and exclusion of strong detrusor overactivity; (5) flexible cystoscopy demonstrating sphincter dysfunction and excluding stenosis of the urethra or vesicourethral anastomosis; and (6) absence of other devices in the urethra aimed at resolving SUI (this point was added after revision of protocol on February 23, 2023 owing to an adverse event occurring in a male patient, see the Results section).
Patients were excluded if they were not self-sufficient, if their urine culture was positive for urinary tract infection (UTI), if the use of magnetic fields was forbidden (i.e., patients with pace-makers), or if they were allergic to the sphincter’s materials (mainly titanium, nitinol, polydimethylsiloxane, and neodymium magnets).
During the screening visit, a thorough anamnesis was collected to determine whether the patient could be enrolled, and dedicated exams were performed (urine culture, urodynamic testing, and flexible cystoscopy). Then, after providing written informed consent, the patient was hospitalized and the proposed device implanted in the operating room through a rigid resectoscope under locoregional anesthesia.
The proposed endourethral device had a diameter of 7 mm and an overall length of 33 mm (for male patients) or 27 mm (for female patients). It consisted of an external case in titanium grade 5, proximal and distal stents, both made of Nitinol. The stents aimed at preventing the device migration into the bladder or outside the body (more details in Supplementary Data S1 and S2). Other components were a unidirectional polymeric valve made of polydimetilsyloxane and a magnetic safety system. The safety system was based on a safety cursor made of titanium grade 5 and two internal magnets (a mobile and a fixed one, NdFeB N52 with a coating in Zn-parylene). Finally, a spring (Phynox) was also included in the magnetic safety system.
The proposed device was featured by two opening pressure levels 18 : in the “open condition” the valve opening pressure was 6.5 ± 0.9 kPa, and it depended only on the intrinsic features of the polymeric valve. In the “closed condition,” the valve hydraulic resistance was enhanced through the magnetic safety system, reaching an opening pressure of 12.0 ± 1.1 kPa. To switch between the two conditions, the patient approached an external permanent magnet to the perianal area. The device could also work by exploiting the “open condition” only, without the need to use the external magnet. In fact, in such a case (“open condition”), the valve still had a hydraulic resistance (6.5 ± 0.9 kPa) that prevented urine flow during standard daily life activities. Once opened (e.g., through the pressure exerted by abdominal muscle contraction), the valve flow resistance reduced, and the patient was able to urinate at low pressure, similar to the physiological one during micturition.
The insertion procedure consisted of the following steps: (1) the urologist tied a suture wire around the distal stent of the device; (2) the 26 Fr resectoscope (item no. 27050SLk, KARL STORZ SE & Co.) was inserted in the urethra under locoregional anesthesia. In a few cases (female patients who benefited from an easier implantation and removal procedure), local anesthesia was used; (3) the resectoscope advanced toward the bladder neck area using an optical vision apparatus; (4) the urologist removed the optical vision apparatus and the internal resectoscope tools, keeping only the external sleeve; (5) the device was inserted into the sleeve lumen; (6) the urologist pushed the device ahead by using a standard endoscopic clamp or a pusher, moving it toward the distal end of the resectoscope, at the bladder neck area; (7) the device exited the resectoscope allowing the enlargement of the top stent inside the bladder, thanks to its shape-memory properties; (8) the urologist removed the resectoscope keeping the device in place; (9) the urologist slightly moved the device to properly position it at the level of the bladder neck. This was achieved by pulling the suture wire tied around the distal stent; and (10) finally, the suture wire was removed and the correct device position was double-checked by means of ultrasound imaging or other imaging techniques (e.g., fluoroscopy) based on urologist’s preference (Fig. 1).
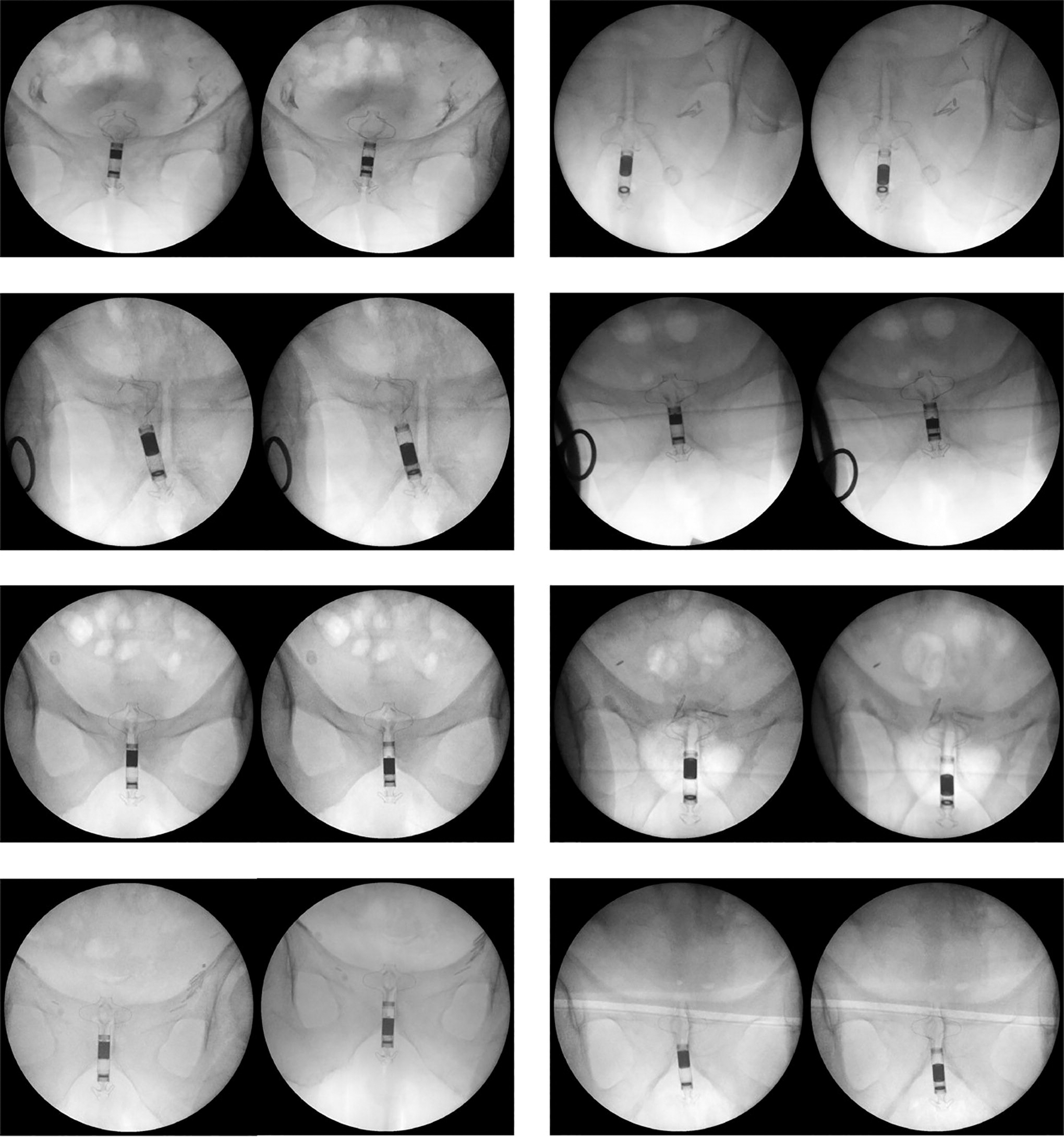
Fluoroscopy images to verify the correct positioning of the device and the activation mechanism: the device is implanted in the “closed condition” (left image) and, after approaching the external magnet to the perianal area, it switches to the “open condition” (right image). Only metal parts are visible. The polymeric valve is transparent to fluoroscopy. The figure shows images for patients IT031, IT032, IT033, IT035, IT037, IT038, IT039, and IT0311.
In total, 10, 30, 60, and 90 days after implantation, the International Consultation on Incontinence Questionnaire on Urinary Incontinence-Short Form (ICI-Q) and a self-assessment questionnaire on the quality of life were administered to the patients to evaluate continence level, as well as the efficacy, acceptability, and usability of the device. The patients were discharged from hospital with the device in “open condition.” In addition to the implanted device, when leaving the hospital, the patients that underwent the first slot of implantation were provided with a suitable external magnet to switch between the “open condition” and the “closed condition.” Instead, for the patients that underwent the second slot of implantation, the external magnet was provided only if the patient was not satisfied with the continence level reached, after the first 10 days (without the external magnet, the device worked in “open condition”). On day 10, 30, 60, and 90, an ultrasound scan of the bladder was performed to verify that the device was correctly positioned and all patients were asked to carry out a 24-hour pad test (mean value obtained from three consecutive days) to objectively assess the urinary continence level. The patients were also suggested to fill out a daily diary about possible side effects or other issues (i.e., in addition to ICI-Q). After up to 90 days, the patient underwent a new cystoscopy to remove the device. In particular, the device was easily removed through the following steps: (1) the resectoscope was inserted into the urethra under locoregional or, in a few cases, local anesthesia; (2) the urologist verified the device position through a camera integrated into the resectoscope; (3) the urologist grasped the distal stent by using forceps and pulled out the device into the resectoscope; (4) when the device was completely inserted into the resectoscope, the urologist removed the resectoscope from the uretral lumen, thus completing the removal procedure.
All possible complications were evaluated using the Clavien-Dindo classification system and recorded. Finally, 21 days after the device removal, a self-assessment questionnaire was administered to evaluate some possible chronic side effects. Finally, a custom checklist questionnaire was filled out by clinicians to assess some key aspects (more details are reported in the Results section).
A normality test (Shapiro–Wilk) was performed on all experimental data, resulting in a normal distribution. A one-way ANOVA test was applied to data, followed by a Tukey test for post hoc comparisons. Statistical analyses were carried out using Prism (v 9.1.1). The significance threshold was set at 5% (*p < 0.05).
Results
The proposed device was successfully implanted and explanted in 8 out of 10 patients (six men, two women, Fig. 1). The average time needed for the implantation and removal procedures was ∼10 minutes and ∼6 minutes, respectively. The results of pad-test, ICI-Q, and a custom checklist are reported in Figures 1 and 2 and Tables 1–3.
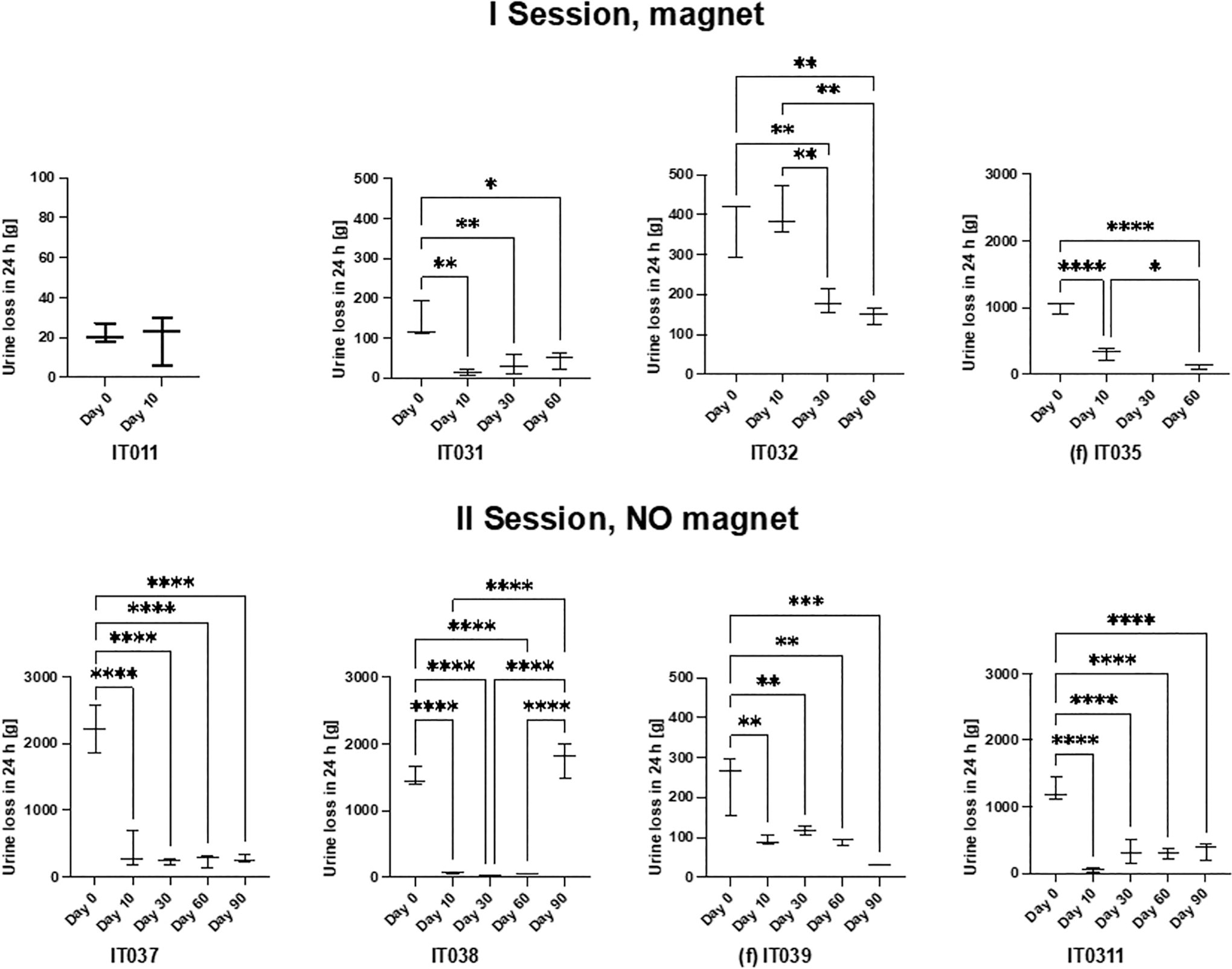
Results of the 24 hours pad-test for the patients in which the device was successfully implanted. In the first session (top), the magnet was used by the patients, whereas in the second session (bottom), patients did not use the magnet and controlled the valve opening by simply exerting an abdominal pressure, then the valve automatically closed after urination ended. The female patients are IT035 and IT039 (f). *=p < 0.05, **=p < 0.01, ***=p < 0.001, ****=p < 0.0001.
Distribution of the Cumulative 24 Hours Pad-Test Results (considering Values from All Time-Points Together) for the Patients in which the Device was Successfully Implanted
M = male patients; F = female patients
Results of ICI-Q in the Patients in which the Device was Successfully Implanted and Explanted
Note: The Value “% ICI-Q Xday” Indicates the Variation of the Total Score of ICI-Q at Day x with Respect to the Initial Condition (a Reduction in the ICI-Q Score Corresponds to an Improvement in the Quality of Life).
ICI-Q = International Consultation on Incontinence Questionnarie-Short Form.
Results of a custom checklist Questionnaire That was Filled out by Clinicians in Order to Evaluate Some Key Aspects
Note: The checklist was based on a custom scoring system. Each question was filled out for each patient and then averaged over the total number of patients. The final result was normalized (10 corresponds to the maximum possible score). The study was considered satisfactory if the score resulted larger or equal to a minimum threshold (defined internally), reported in a column of the table.
The study yielded excellent results, particularly when considering the initial UI severity (Fig. 2). For example, IT011 was a male patient who could be classified as having mild incontinence rather than severe one. In his case, no dramatic improvements were registered (median value: −30% of urine loss with respect to the initial condition Fig. 2). Consequently, he requested to remove the device after 50 days. However, for the other patients, as the degree of incontinence was moderate to severe, the device successfully restored a high level of continence (Fig. 2). This emerges from the analysis of the median values (see Table 1 for all details). Looking at the patients with moderate-to-severe UI, the median values of continence restoration ranged from 59% to 99% (Figs. 2 and 3a,b).
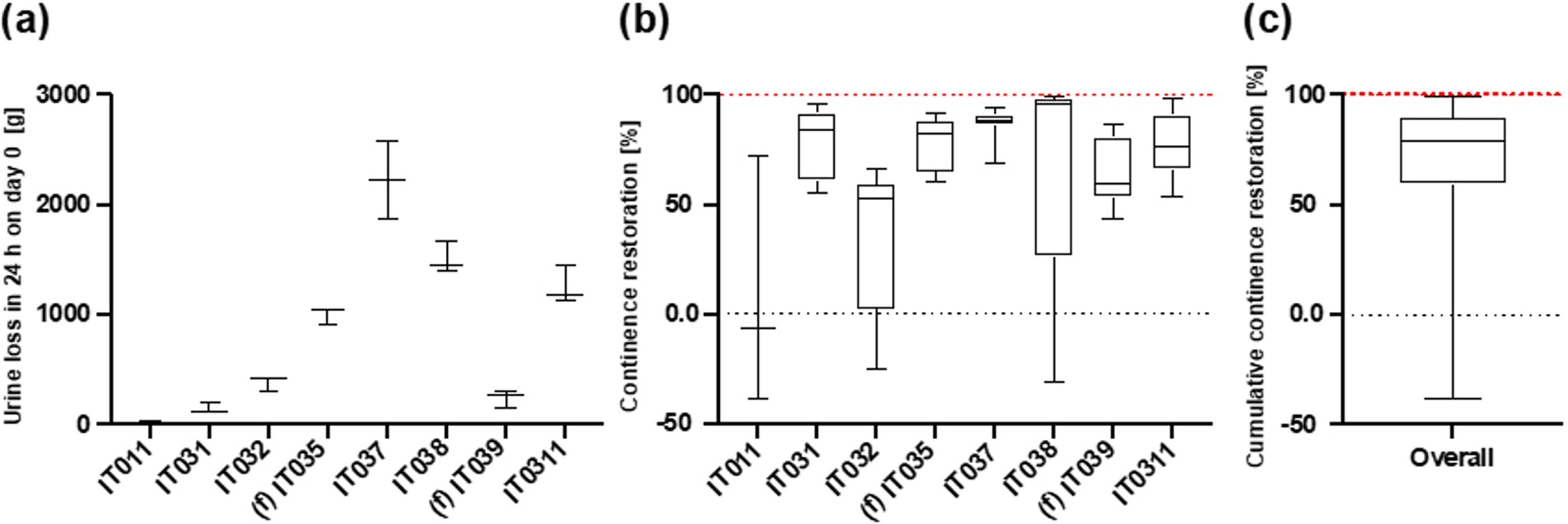
Incontinence level and continence restoration for the patients in which the device was successfully implanted [female patients are IT035 and IT039 (f)].
When considering the cumulative continence restoration, we observed that the 25% percentile value corresponded to 59% of continence restoration (Fig. 3c). This means that at least 75% of the patients experienced a substantial improvement (>50%) in their continence level. The median value was 82%, representing the middle point of data distribution, indicating a significant shift toward continence restoration for the majority of patients.
The 75% percentile value of 91% of continence restoration further supports the trend of significant continence improvement in many cases. The maximum value was 0.9935, implying an almost complete continence restoration achieved.
In summary, for the patients in which the device was successfully implanted and explanted, the statistical analysis of data underscores that the device efficacy in restoring continence is most pronounced in patients with moderate-to-severe UI, whereas those affected by mild UI experienced only a slight improvement. This observation reinforces the potential of the device to offer substantial relief to patients with severe forms of UI.
Remarkably, the restoration of continence levels was excellent also for patients who did not use the external magnet, and who left the device for the whole 3-month period in the “open condition” (see Fig. 2, bottom: II session: no magnet). This means that the hydraulic resistance of the valve, even without activating the magnetic system, was enough to guarantee a satisfying continence restoration level.
The study was not successfully completed in 2 out of 10 patients (both men) owing to adverse events. Only a severe adverse event (i.e., urinary retention) occurred in one patient due to the presence of a previously implanted device to treat UI (i.e., an obstructor). In the other patient it was not possible to implant the device due to bleeding occurred during the implantation procedure.
Among the implanted devices, only one stopped working 76 days after the implantation: in this case, the valve remained completely open and brought to incontinence—see data for patient IT038 in Figure 1, on day 90). The majority of patients (7 out of 10 reported good tolerability of the device (see Table 3 question 4) and very good results about continence, as well as a remarkable improvement in the quality of life (Table 2). Only one of the eight patients that successfully received the implant required to remove the device before the end of the study (on day 50). This was the patient (IT011) who, as already mentioned, was affected by mild UI and consequently did not observe remarkable improvements. Migration of the device into the bladder or along the urethra, which was the main issue observed in the previous pilot study, did not occur in any patient, in the course of this study.
Device removal was easily performed in all patients (average time needed: 6 minutes) and the initial UI status was restored after device removal. No complications or UTIs were reported during the 90 days or after the explant (as demonstrated by the results obtained at the follow-up, carried out 21 days after the device removal).
Discussion and Conclusion
Patients undergoing radical prostatectomy might experience UI primarily because of harm to the lower part of the urethral sphincter, resulting in SUI. Concerning men, the AUS is presently regarded as the most effective treatment for UI in such cases. 4,9 –11 Usually, the AUS is made of several components, including a cuff that surrounds the urethra, a pressure-regulating balloon reservoir, and a control pump placed within the scrotum. The cuff is surgically implanted around the urethra to simulate the function of the natural sphincter muscles, allowing the patient to control the urine flow voluntarily. The reservoir holds a saline solution, and the pump is used to deflate or inflate the cuff, enabling the user to start or stop the flow of urine. However, the implantation of these devices is featured by a high invasiveness and relatively high rate of related complications. The AMS 800 continues to be considered the benchmark, yet it lacks the capability to reposition the cuff in instances of postoperative urethral atrophy and does not offer the possibility to modify the device’s pressure once it has been activated. In this study, we propose an alternative endourethral magnetic sphincter, characterized by a more straightforward and less invasive implantation procedure and a short learning curve, which could be offered to patients suffering from SUI irrespectively of their sex. We enrolled 10 patients (eight men and two women) who reached the defined endopoints. The results show that the implantation was successfully accomplished in eight of them (six men and two women), with excellent results in terms of continence restoration and quality of life improvement.
One of the notable advantages of the AUS is the ability to provide on-demand urination control, allowing users to empty their bladder at their convenience. This feature significantly enhances the patient’s ability to engage in daily activities without the fear of urinary losses, thereby improving the overall quality of life. To do so, state-of-the-art devices use complex and invasive means (e.g., manual pumps 12 remotely controlled using hydraulic or pneumatic systems or inflatable balloons 21 ). The device described in this study, compared with the conservative solutions typically employed, 4 showed many advantages in terms of therapy efficacy. In addition, the device showed significantly lower invasiveness compared with other nonconservative therapeutic devices, especially extraurethral devices, which usually require invasive surgical procedures to implant them and, consequently, complex surgeries for removal. We demonstrated ease of implantation and explant, ease to control opening, and closure of the device especially without the use of magnetic activation/deactivation (patients that did not use the magnet controlled the opening and closure of the valve simply by exerting an abdominal pressure in a more straightforward way). This allowed to restore urinary continence up to excellent levels (mean urine’s losses between −62% and −99%) and with good tolerability (no patients reported pain; only one patient reported discomfort, whereas seven patients reported no pain and just slight discomfort in the first days after implantation). Seven out of eight patients discharged with the device into the body terminated the study until day 90 with the device that properly worked. In our cohort, only one device deficiency occurred 76 days postimplantation owing to a mechanical failure of the internal valve (i.e., the mobile parts of the valve remained in a fully open conFig.uration), thus constituting a mild adverse event (i.e., UI). Only a severe adverse event (i.e., urinary retention) occurred in one patient, but it was owing to a failure to comply with inclusion/exclusion criteria (that patient had a previously implanted device in the urethra). Only for one patient, the implantation was not possible owing to bleeding during the procedure. None of these patients required an urgent intervention and no UTIs were reported during the study. Postoperative observation reported the emergence of dysuria that was controlled by analgesics. The quality of life of patients considerably improved in the majority of the cases with a reduction of urine losses impact between 51% and 99%. Only one patient reported no variations in the quality of life. Device removal was easily performed in all patients.
After removing the device, the initial UI condition was restored in all patients. Questionnaires filled out 21 days after device removal demonstrate that no chronic effects or changes in the quality of life were caused by the device implantation in comparison with the initial condition. Furthermore, no chronic discomfort or pain, no irritation effects, or changes in urination frequency were reported. Also, sexual activity was not modified with respect to the initial condition.
In conclusion, this pilot study on 10 patients demonstrated the safety, usability, and efficacy of an endourethral artificial sphincter to face UI. Future perspectives include the execution of a large-scale clinical study and certification of the device, which look promising for driving a drastic reduction in the psychological burden typically associated with UI and a considerable reduction of health care systems’ costs.
Footnotes
Acknowledgment
This work was supported by Relief srl, an Italian startup, Spin-off of Scuola Superiore Sant’Anna (www.reliefsrl.com).
Authors’ Contributions
M.C., F.P., I.P., F.M., and N.P.: Main principal investigators of the pilot study and performed the clinical evaluation on humans. G.L., T.M., L.M., L.R.: Given substantial contributions to the conception or the design of the article, to acquisition, analysis, and interpretation of the data. G.P., R.M.S., and R.P.: Revised it critically. All authors have participated to drafting the article. All authors read and approved the final version of the article.
Author Disclosure Statement
The authors certify that although there is a conflict of interest for some authors regarding the protocol, who are members of the company supporting the study, all analyses, results, and conclusions were fairly and thoroughly reported and were not influenced in any way by this conflict of interest.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Data S1
Supplementary Data S2
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.