
Abstract
We studied the sensitivity of polymerase chain reaction (PCR) for detecting DNA of migratory larvae of Trichinella spiralis at an early stage of infection with this parasite. We derived primers for PCR from a 1.6-kb repetitive sequence of the genome of T. spiralis and used PCR to detect Trichinella-specific DNA in blood of mice infected with 20, 100, or 300 muscle-derived larvae of T. spiralis at 3–21 days postinfection (dpi). We detected T. spiralis DNA in blood of mice infected with 20 larvae at 5 and 6 dpi, with a detection rate of 7.69% and in blood of mice infected with 100 larvae at 5–12 dpi, with a peak detection rate of 38.46% at 7 dpi. PCR detected T. spiralis larvae at 5–17 dpi in mice infected with 300 larvae, with detection rates exceeding 50% from 5 to 10 dpi and a peak rate of 61.54% at 7 dpi. The detection rates of T. spiralis larvae with PCR in the three groups of mice showed an increasing trend with an increase in the infecting dose of larval parasites (F = 17.811, p < 0.01). Our findings indicate that the sensitivity of PCR for detecting DNA migratory larvae of T. spiralis in blood of mice infected with this parasite depends on the severity of infection and the time elapsed after infection, and suggest that PCR may be useful for detecting Trichinella infection at an early stage in humans and food animals that test negatively for anti-Trichinella antibodies.
Introduction
T
Female adult T. spiralis parasites produce live offspring at 5 days postinfection (dpi) (Denham and Martinez, 1970), and one female worm produces about 1500 larvae in 5–10 days, with the production of larvae having the potential to continue for as long as the female worm remains in the intestine. The duration of production of T. spiralis larvae differs in different hosts, persisting for 10–20 days in mice and rats, and 4–6 weeks in humans (Campbell, 1983; Bell, 1998). The newly hatched larvae migrate to bloodstream via the lymphatics and veins, and then invade skeletal muscle cells to give rise to the complex of infective L1 larvae and the nurse cells that evolve from invasion of these muscle cells by the larvae. However, a proportion of newly hatched T. spiralis larvae recirculate within the vasculature for several hours, and a smaller proportion extravasate but can re-enter the circulatory system via the lymphatics (Wang and Bell, 1986). The detection of circulating Trichinella larvae or their products may be useful for the early diagnosis of trichinellosis. Polymerase chain reaction (PCR) has been widely applied to the amplification of DNA from muscle larvae of T. spiralis (Pozio and Murrell, 2006), and the detection through PCR of larvae of the organism in blood or muscle of hosts has been reported (Uparanukraw and Morakote, 1997; Caballero-Garcia and Jimenez-Cardoso, 2001; Atterby et al., 2009), but the effects on such detection of the infectious dose of Trichinella parasites and of the duration of the postinfection period have not been ascertained. We therefore studied the early detection by PCR of migratory larval DNA of T. spiralis in blood of mice experimentally infected with 20, 100, and 300 of these parasites from 3 to 21 dpi.
Materials and Methods
Trichinella spp. and experimental animals
The Trichinella isolates used in the study were T. nativa (ISS10), T. britovi (ISS100), T. pseudospiralis (ISS13), and T. nelsoni (ISS29), all of which were obtained from the Trichinella Reference Center (IRC; Rome, Italy), and T. spiralis (ISS534), obtained from domestic pigs in Nanyang city of Henan Province, China. All of these Trichinella isolates were maintained by serial passage in Sprague–Dawley rats at 6- to 8-month intervals in our department (e.g., Department of Parasitology, Medical College, Zhengzhou University). Forty-day-old specific pathogen-free (SPF) Kunming mice weighing 20–25 g were purchased from the Experimental Animal Center of Henan province and bred in plastic microisolator cages.
Infection of mice and collection of blood
Thirty-nine male SPF Kunming mice were divided into three groups of 13 mice each, which were, respectively, infected with 20, 100, and 300 muscle larvae of T. spiralis. From 3 to 21 dpi, 50 μL of tail blood was collected daily from each animal into acid citrate dextrose. The infected mice were euthanized, skinned, and eviscerated at 40 dpi. The carcasses were minced and digested for 4 hours at 43°C in a solution of 0.33% pepsin (1:31,000) and 1% HCl. At the end of the digestion process, the entire digests from the beaker are poured through a # 80 sieve (180 μm mesh) into a 2 L separatory funnel, and the beaker and sieve were rinsed with another 100 mL of warm (37°C) water, and settled for 30 minutes. About 40 mL of the fluid is drained from the funnel into a 50 mL glass centrifuge tube and settled another 10 minutes; then, all but 10 mL of bottom fluid is aspirated out. The final, clarified 10 mL of sediment is poured into a gridded Petri dish and examined for Trichinella larvae with an Olympus microscope (Tokyo, Japan) (Gamble et al., 2000).
DNA extraction
Twenty muscle larvae of each of T. spiralis, T. nativa, T. britovi, T. pseudospiralis, and T. nelsoni were, respectively, collected from the experimentally infected SPF Kunming mice in our department under an Olympus microscope and treated at 55°C for 12 hours with 100 μL of cell lysis buffer (0.1 mg/mL proteinase K–1% sodium dodecylsulfate). Larval DNA of each of five Trichinella species was obtained according to the classic phenol–chloroform extraction method (Connolly et al., 1995). This was done by first gently mixing the lysate obtained from each lysis-buffer-treated specimen of the five Trichinella species larvae with an equal volume (400 μL) of a phenol–chloroform (1:1, v/v) mixture, and centrifuging the resulting mixture at 4500 g for 6 minutes. About 350 μL of supernatant was then transferred to a fresh centrifuge tube and mixed with an equal volume of a mixture of chloroform–isoamyl alcohol (24:1, v/v). The resulting mixture was then again centrifuged for 6 minutes at 4500 g. The upper phase of the supernatant was transferred to a centrifuge tube, and two volumes of ethanol were added to precipitate the Trichinella larval DNA at −20°C in the presence of 0.2 M NaCl for 4 hours. After centrifugation of this precipitate at 5500 g for 10 minutes, the supernatant was removed and 1 mL of 70% precooled ethanol was added to the remaining pellet and centrifuged at 5500 g for 5 minutes, and the supernant was discarded. The pellet was then air-dried and was dissolved and diluted in 40 μL of double-distilled water. The quantities of DNA of each of the five Trichinella species muscle larvae obtained in this manner and used as templates for PCR were equivalent to DNA of 20, 1, 0.04, and 0.02 larvae.
DNA of 1, 2, and 3 muscle larvae of T. spiralis was extracted as described by Bandi et al. (1993). In brief, the larvae were transferred under an microscope into 100 μL of cell lysis buffer and incubated overnight at 55°C. DNA was obtained from the larvae by phenol–chloroform extraction and ethanol precipitation using the above-described method, with the resulting whole extracts used as templates for PCR.
Fifty microliters of normal mouse tail vein blood containing 1, 2, or 3 T. spiralis larvae, or 50 μL of tail vein blood from a mouse experimentally infected with 20, 100, and 300 T. spiralis muscle larvae during 3–21 dpi was homogenized with 200 μL of red blood cell lysis buffer (155 mM NH4Cl, 10 mM KHCO3, and 0.1 mM ethylenediamine tetraacetic acid; pH 8.0), incubated at room temperature for 10 minutes, and centrifuged at 400 g for 10 minutes, with the sediment then being rinsed twice with 400 μL of red blood cell lysis buffer. The supernatant was decanted after centrifugation (Dupouy-Camet et al., 1993). The resulting white blood cell pellets were then treated overnight at 55°C with 100 μL of cell lysis buffer, and T. spiralis DNA was isolated from each pellet through the phenol–chloroform method. Blood of uninfected normal mice was used as control.
Polymerase chain reaction
Primers were derived from a 1.6-kb repetitive sequence of the T. spiralis genome (deVos et al., 1988) and had the following sequences: 5′-CTTGTAAAGCGGTGGTGCGTA-3′ and 5′-CATAGAGAGGCAACATTACCT-3′ (Dick et al., 1992). PCR was performed in 25 μL of a solution of 1 × PCR buffer (TaKaRa Biotechnology Co. Ltd., Dalian City, China), 150 mM MgCl2, 0.2 mM deoxynucleoside triphosphates, 1 unit of Taq DNA polymerase (TaKaRa Biotechnology Co. Ltd., Dalian City, China), and 0.4 μM primer. Amplification was done in 35 cycles each of 1 minute at 94°C, 1 minute at 54°C, and 1 minute at 72°C. The PCR products were detected using 2% agarose gel electrophoresis at 100 V and 63 mA for 40 minutes, and the gel was stained with GeneFinder (Bio-V Bioscientific Co. Ltd., Xiamen City, China).
Statistical analysis
All statistical analyses were done with SPSS for Windows version 13.0 (SPSS Inc., Chicago, IL), with the comparisons of variables made through nonparametric tests, comparison of correlation coefficients, and trend analysis. χ 2 analysis, analysis of variable with multiple comparisons (analysis of variance), and comparison of coefficients of correlation were used in this study. All statistical tests were considered significant at a p-value of 0.05.
Results
Products and sensitivity of PCR for Trichinella spp.
A single specific band of DNA of 510 bp and two unexpected bands of DNA of 670 and 1200 bp, respectively, were amplified by PCR of DNA extracted from 20 muscle larvae of T. spiralis, T. pseudospiralis, and T. nelsoni using the primers described above (Fig. 1). Lane 1 in Figure 1 was the positive control template corresponding to DNA of 0.02 muscle larvae of T. spiralis and three fragments (510, 670, and 1200 bp) were amplified. The templates that produced amplification corresponded to DNA of 1, 0.04, and 0.02 muscle larvae of these Trichinella species; however, PCR failed to amplify DNA extracted from the muscle larvae of T. nativa or T. britovi. The PCR product of DNA corresponding to 0.04 and 0.02 larvae of T. pseudospiralis consisted only of the 510-bp band of DNA, whereas DNA corresponding to one whole larva of this organism yielded three DNA fragments (510, 670, and 1200 bp).

Polymerase chain reaction (PCR) detection of migratory larvae in tail blood of mice infected with 300 muscle larvae of Trichinella spiralis during 3–9 days postinfection (dpi) (
Sensitivity of PCR detecting 1 to 3 muscle larvae of Trichinella
Amplification by PCR of DNA extracted from 1 to 3 T. spiralis larvae and of DNA from 1 to 3 T. spiralis larvae in 50 μL of normal mouse blood yielded three fragments of DNA (510, 670, and 1200 bp), but did not yield any Trichinella-specific DNA from blood of uninfected mice.
PCR detection of tail blood of mice infected with T. spiralis larvae
The results of PCR for detecting migratory larval DNA in the tail blood of mice infected with 20, 100, and 300 larvae of the parasite are shown in Figures 1 and 2.
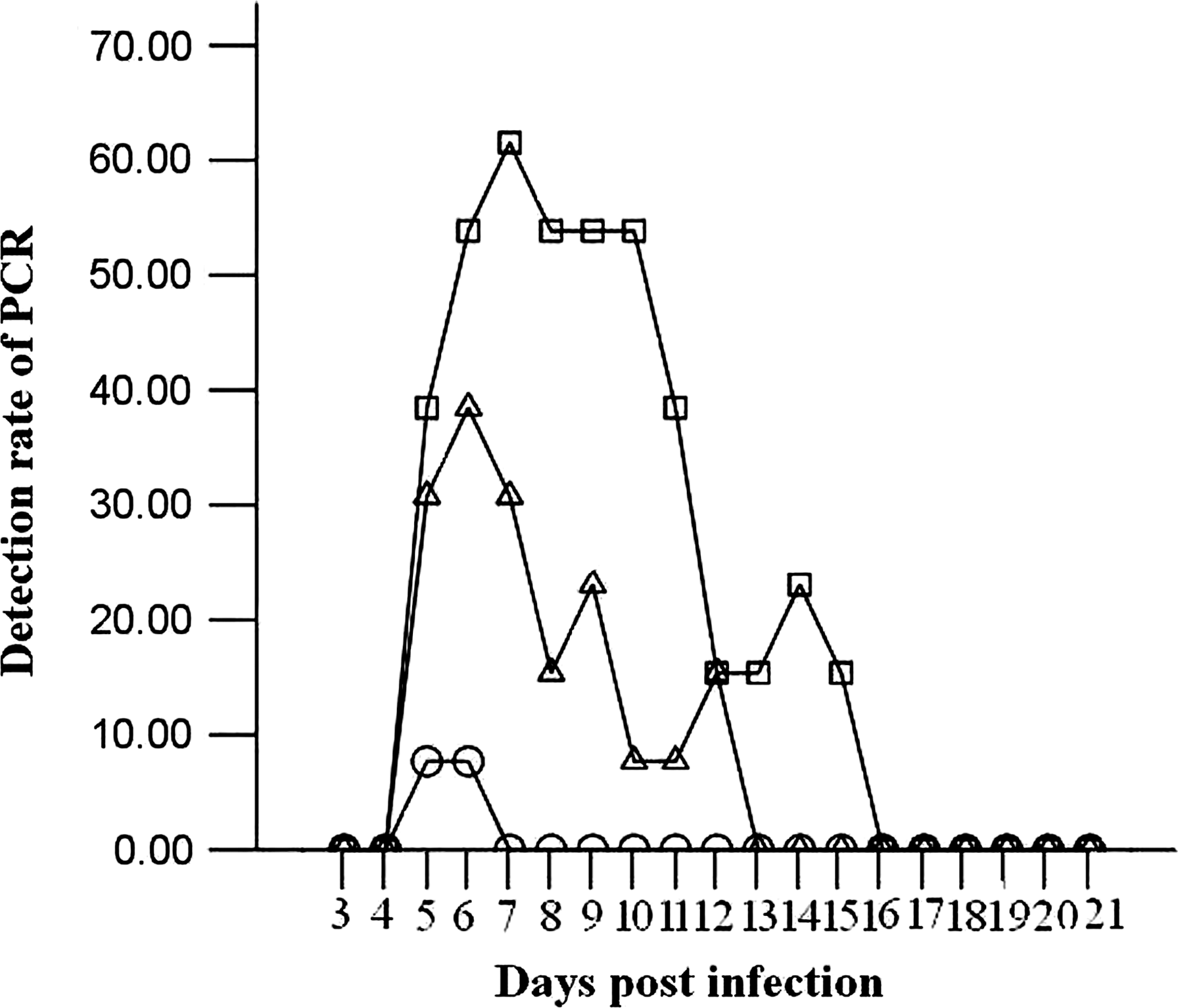
The positive rate of PCR detection of migratory larvae in tail blood of mice infected with 20 (○), 100 (△), and 300 (□) muscle larvae of T. spiralis during 3–21 dpi.
T. spiralis DNA was not detected in blood of mice infected with 300 larvae at 3–4 dpi (lanes 2 and 3 in Fig. 1A) and from 16 dpi (lane 8 in Fig. 1B) to 21 dpi (Fig. 2).
The amplification product of PCR done in tail blood of mice infected with 300 T. spiralis larvae was a 510-bp DNA fragment from 5 to 15 dpi (Fig. 1). The mean ± standard deviation of parasitic burdens of mice infected with 20, 100, and 300 T. spiralis larvae at 40 dpi was 481.85 ± 45.60, 3247.25 ± 406.99, and 6279.72 ± 443.54 larvae/gram (l pg) of muscle, respectively. T. spiralis DNA was detected in blood of mice infected with 20 larvae at 5 and 6 dpi, at a rate of 7.69% (1 of 13 specimens) on both days (shown as –○– in Fig. 2), and from 5 to 12 dpi in blood of mice infected with 100 larvae (shown as –Δ– in Fig. 2), with a peak positive rate of 38.46% (5 of 13 specimens) at 6 dpi being the highest during the detection period. PCR detected T. spiralis DNA in blood of mice infected with 300 larvae from 5 to 15 dpi, with peak detection rates of 53.85% (7 of 13 specimens) at 6 dpi and from 8 to 10 dpi, and 61.54% (8 of 13 specimens) at 7 dpi (shown as –□– in Fig. 2). The difference in the rate of PCR positivity among mice infected with 20, 100, and 300 larvae was statistically significant (χ 2 = 12.253, p < 0.01), and the rates of PCR positivity in the three groups of mice were significantly correlated with infecting doses of T. spiralis larvae (r = 0.497, p < 0.01) and showed an increasing trend with an increasing infecting dose (F = 17.811, p < 0.01). PCR positivity on different days after infection was significantly positively correlated with the period after infection in mice infected with 100 and 300 larvae (r 100 = 0.394, p < 0.01; r 300 = 0.456, p < 0.01).
Discussion
The 1.6-kb sequence of the T. spiralis genome from which we derived the PCR primers for this study was initially thought to be specific for a porcine isolate of T. spiralis, and has a copy number of 2800 per haploid genome of the organism, with tandem organization (Klassen et al., 1986a, 1986b). Because of the high copy number of this sequence, PCR based on the primers derived from the sequence is very sensitive to this 1.6-kb fragment of T. spiralis DNA. As a result, the PCR-based amplification of DNA equivalent to 0.02 muscle larvae of T. spiralis is efficient. Two unexpected bands of DNA, 570 and 1200 bp, respectively, were observed as a consequence of the presence of a 1.9 kb variant of the 1.6 kb repetitive sequence and of polymorphism of the T. spiralis genome. Similar fragments were obtained from T. spiralis and T. nelsoni when their muscle larval DNA was amplified with multiple pairs of the primers derived from the 1.6-kb sequence (Soulé et al., 1993), but PCR failed to amplify DNA of T. pseudospiralis. The difference may be related to the different primers used in our study. Southern blot analysis showed that DNA of T. pseudospiralis hybridized only weakly with DNA probes derived from the 1.6-kb sequence of T. spiralis, indicating partial homology of the genomes of T. spiralis and T. pseudospiralis. T. pseudospiralis in domestic and sylvatic animals has been reported in Asia, Australia, Europe, and North America (Pozio and Murrell, 2006), and three outbreaks, involving a total of 92 cases of human trichinellosis caused by T. pseudospiralis, have been documented in Kamchatka, Thailand, and France (Jongwutiwes et al., 1998; Ranque et al., 2000). T. nelsoni is an etiologic agent of sylvatic trichinellosis and is distributed in eastern Africa, from Kenya to South Africa, with about 100 cases of human infection by T. nelsoni having been reported in Kenya and Tanzania (Pozio, 2005). Our results showed that PCR was useful for detecting human and animal infection caused by T. spiralis, T. pseudospiralis, and T. nelsoni. This suggests that infection by all species of Trichinella could potentially be identified by a simple multiplex PCR (Zarlenga et al., 2001). Trichinellosis caused by all species of Trichinella can effectively be treated with abendazole or mebendazole (Dupouy-Camet et al., 2002).
Female adult Trichinella worms are thought to give birth to larvae throughout their lives in hosts, with the newborn larvae entering the circulating blood of the host and becoming distributed throughout host's body. However, it is only in skeletal muscles that the larvae reach the infective stage and become encapsulated at 26 dpi. Before collagen capsules of the larvae are formed, biopsy is not a reliable method for diagnosing trichinellosis. The disadvantage of serologic methods (such as ELISA using excretory–secretory antigens) for identifying Trichinella infection is that they give false-negative results during the early stages of infection. In mildly or moderately infected animals, a lag occurs between the time at which larvae become infective in a new host and the occurrence of positive serologic results for trichinellosis (Gamble et al., 2004). This delay in the production of antibodies to Trichinella means that animals infected with smaller numbers of Trichinella larvae may not be identified for several weeks after they become parasitized, even in the presence of actively infectious muscle larvae (Gajadhar et al., 2009). The method of artificial digestion recommended by the International Commission on Trichinellosis for the control of Trichinella in domestic and wild animals intended for human consumption cannot be used to detect infective preencapsulated larvae in meat because these larvae are not resistant to the enzymatic digestion used in this method, and some dead larvae are not recovered after passage through the sieves and sedimentation used in the method, with the result that it yields false-negative results during the early phases of parasitic infection. This window of false-negativity of both serologic tests and the method of artificial digestion prevents the use of these techniques for both the pre- and postmortem surveillance of food animals for Trichinella infection. The use of PCR for the early detection of migratory or preencapsulated Trichinella larvae in blood or meat of such animals may therefore be an effective means of preventing human trichinellosis, as well as for its early diagnosis.
Although PCR has been used to detect migrating T. spiralis larvae in blood of experimentally infected mice (Uparanukraw and Morakote, 1997; Caballero-Garcia and Jimenez-Cardoso, 2001), no detailed data have been reported about the results of PCR in detecting T. spiralis at different infective doses in mice, or about the sensitivity of PCR for detecting the organism in the period after infection, particularly in the case of mild infection. Considering that PCR may be an effective diagnostic tool for trichinellosis, we infected mice with various doses of T. spiralis, of which 20 larvae are thought to be the smallest infective dose in mice (el-Shazly et al., 2002), and collected blood daily from 3 to 21 dpi to better assess the efficacy of PCR for the early diagnosis of trichinellosis.
The stage of larval deposition by T. spiralis in infected mice generally begins within 5 dpi and continues for the next 10–20 days. Hence, our detection of Trichinella DNA from 5 to 15 dpi corresponds to the period in which newborn larvae of the organism migrate through blood. An earlier study had found that PCR detected circulating T. spiralis larvae in the tail blood of mice infected with 300–350 larvae from 5 to 14 dpi (Uparanukraw and Morakote, 1997), which was similar to our finding in mice infected with 300 larvae. However, we also detected Trichinella DNA at 15 dpi, as well as examining more mice and using a different method of DNA extraction than in this earlier study. The identification through PCR and Southern blot analysis of T. spiralis DNA at 3–17 dpi in vena caval blood samples from mice infected with 300 larvae of the organism (Caballero-Garcia and Jimenez-Cardoso, 2001) suggests that Southern blotting may be a more sensitive means of detecting products of PCR for Trichinella DNA than is agarose gel electrophoresis. PCR was not as effective for detecting T. spiralis DNA in human patients with trichinellosis as it was for detecting T. spiralis in mice, even though the larval migratory period of the organism in blood of humans is longer than that in mice. Among 37 cases of human trichinellosis, Trichinella-specific DNA was detected in only two cases at 4–6 weeks after infection (Robert et al., 1996). DNA, IgG, a heme compound copurified with DNA from bloodstains, and other elements in host blood inhibit the sensitivity of PCR for detecting specific DNA, and may contribute to such impaired sensitivity (Akane et al., 1994; Abu Al-Soud and Rådström, 2000; Al-Soud et al., 2000; Al-Soud and Rådström, 2001). In view of this, and of the effect of the relatively short duration of the larval migratory period of T. spiralis on its identification, greater sensitivity is needed for its detection. As compared with routine PCR, real-time PCR is more sensitive in terms of DNA amplification and may provide an advantage in this regard. In a recent study, Atterby et al. (2009) reported positive PCR results for real-time PCR in detecting Trichinella DNA in samples of mouse and fox muscle containing one larva per gram and diluted 100-fold. Because infective Trichinella larvae are expelled through the intestine before they spread extensively through blood, it has been suggested that PCR may be more effective for detecting Trichinella DNA in fecal samples during the first 3 days after infection than for use in detecting DNA of the organism in blood, and PCR has recently been used to detect Trichinella larvae in mouse feces (Golab et al., 2009).
Conclusion
Our study indicates that primers derived from a 1.6-kb repetitive sequence of the T. spiralis genome may be useful for identifying mammalian infection with T. spiralis, T. pseudospiralis, and T. nelsoni. In our study, the diagnostic value of PCR for trichinellosis was dependent on the infecting dose of T. spiralis and was also related to the period after infection when PCR was done. PCR may be useful for the early detection of Trichinella infection in food animals and humans, in whom serologic tests for anti-Trichinella antibodies yield negative results during the early stages of infection, and for immunosuppressed hosts.
Footnotes
Acknowledgments
This work was supported by Grant 2010CB530000 from the National Basic Research Program of China, Grant 30972492 from the National Natural Science Foundation of China, and Grant 2008-145 from the Major Public Research Project of Henan Province.
Disclosure Statement
No competing financial interests exist.