
Abstract
Multilocus sequence typing (MLST) was used to examine the clonal relationship and genetic diversity of 71 Vibrio parahaemolyticus isolates from clinical and seafood-related sources in southeastern Chinese coast between 2002 and 2009. The tested isolates fell into 61 sequence types (STs). Of 17 clinical isolates, 7 belonged to ST3 of the pandemic clonal complex 3, with 3 strains isolated in 2002. Although there was no apparent clonal relationship found between clinical strains and those from seafood-related sources positive with pathogenic markers, there were clonal relationships between clinical strains from this study and those from environmental sources in other parts of China. Phylogenetic analysis showed that strains of 112 STs (61 STs from this study and 51 retrieved from PUBMLST database covering different continents) could be divided into four branches. The vast majority of our isolates and those from other countries were genetically diverse and clustered into two major branches of mixed distribution (of geographic origins and sample sources), whereas five STs representing six isolates split as two minor branches because of divergence of their recA genes, which had 80%–82% nucleotide identity to typical V. parahaemolyticus strains and 73.3%–76.9% identity to the CDS24 of a Vibrio sp. plasmid p23023, indicating that the recA gene might have recombined by lateral gene transfer. This was further supported by a high ratio of recombination to mutation (3.038) for recA. In conclusion, MLST with fully extractable database is a powerful system for analysis of clonal relationship for strains of a particular region in a national or global scale as well as between clinical and environmental or food-related strains.
Introduction
A number of genetic fingerprinting techniques have been used to examine the relatedness of several bacterial species such as pulse-field gel electrophoresis (PFGE), ribotyping, and multilocus sequence typing (MLST) (Maiden et al., 1998; Kai et al., 2008; Pavlic and Griffiths, 2009). MLST has been found suitable for a variety of bacterial species (Nallapareddy et al., 2002; Chen et al., 2006; Foley et al., 2006). Chowdhury et al. (2004) approached MLST based on four genes on chromosome I for subtyping of pandemic strains of V. parahaemolyticus. Gonzalez-Escalona et al. (2008) used seven house-keeping genes to examine the genetic diversity of V. parahaemolyticus. Sixty-two sequence types (STs) were identified, and three major clonal complexes (CCs) were found, two for isolates from the Pacific and Gulf coasts of the United States and a third one contained strains belonging to the pandemic CC3 (Gonzalez-Escalona et al., 2008). The CC3 strains have worldwide distribution (Okuda et al., 1997; Chiou et al., 2000; DePaola et al., 2003; Martinez-Urtaza et al., 2004, 2005; Nair et al., 2007; Ansaruzzaman et al., 2008).
Several papers reported the sero-diversity and genetic structure of some clinical and environmental isolates of V. parahaemolyticus. Chao et al. (2009) reported that the pandemic clonal serovars of their clinical isolates from 2005–2008 included O3:K6, O1:KUT, O1:K25, O1:K26, and O4:K68, with O3:K6 being the dominant serovar. Yan et al. (2011) used an extended MLST scheme [having four gene loci identical to MLST by Gonzalez-Escalona et al. (2008)] on 174 global strains, including 7 clinical strains between 2003 and 2007 and 33 environmental isolates between 2006 and 2007 from mainland China. They found three major clonal groups corresponding to the groups of pre-1996 old-O3:K6 strains CC2, post-1996 pandemic strains (CC3), and nonclinical isolates (CC5), respectively. One of the CC3 strains in that paper was a clinical strain isolated in the southern Chinese province of Guangxi in 2003. However, genetic diversity and clonal relationship of V. parahaemolyticus strains from southeastern Chinese coast remain uncharacterized over a wider time period. In this study, we used MLST to examine the genetic population structure of selected V. parahaemolyticus strains from clinical and seafood-related sources between 2002 and 2009 and to analyze the clonal relationship of Chinese isolates to those from other parts of the world.
Materials and Methods
Bacterial strains, cultures, and DNA extraction
Seventy-one V. parahaemolyticus isolates in our strain collection (658 strains, mostly from seafood-related samples) from 2002 to 2009 were selected. There were 17 isolates from clinical sources, including 10 from Zhejiang Center for Disease Control and Prevention (Zhejiang CDC), China, and 7 from Hangzhou CDC, Zhejiang, China. Fifty-three isolates were from seafood-related sources (seafood and their environments) in the southeastern Chinese coastal provinces of Zhejiang and Fujian. These seafood-related isolates were selected based on the presence of major or putative virulence genes such as tdh or trh (Vongxay et al., 2008) or those representing type 3 secretion systems (T3SS) or putative T6SS by polymerase chain reaction (PCR) typing (Table 1 and Supplementary Table S1; Supplementary Data are available online at
Ordered as dnaE, gyrB, recA, dtdS, pntA, pyrC, and tnaA.
ND, not done.
PCR amplification
For direct sequencing of the PCR products of seven loci as described in PUBMLST (
Identification of CCs, groups, and singletons
The eBURST program v3.0 (Feil et al., 2004) was used to subdivide 201 STs (representing 386 strains) into groups of related isolates and CCs. These included 61 STs (71 strains) from this study and all 140 STs (315 strains) available from PUBMLST database as of December 2009 (representing strains from Bangladesh, other parts of China, Chile, Ecuador, India, Japan, Korea, Mozambique, Norway, Peru, Spain, Thailand, and United States). Of the 386 strains, 185 were from environmental sources (47.9%). We used the most stringent group definition that STs share identical alleles at six or seven of the MLST loci with at least one other member of the group. If a group contains closely related STs, which have diversified from founding genotype, the group is considered as a CC. Any two STs differing form each other at a single locus were defined as single-locus variant (SLV) and two STs differing form each other at two loci as double-locus variant (DLV). The ST that has the largest number of SLVs in a CC is considered as the founding genotype. The statistical confidences for the founding genotype were assessed using 1000 bootstrap resamplings. STs that do not belong to any groups are called singletons (Feil et al., 2004).
Determination of allelic profiles and STs
The trace files of new alleles were submitted to the curator for verification. For each gene, new alleles were given with arbitrary allelic numbers. Combination of allelic numbers for a particular isolate constituted allelic profile for that isolate. Each unique allelic profile was assigned an ST number by the curator. Information of the allelic profiles and STs can be found on
Genetic diversity and phylogenetic analyses
Genetic diversity statistics were computed on sequences of individual loci or on concatenated sequences of each isolate by DNASP v5.0 (Librado et al., 2009). The statistics include the number of polymorphic sites, percentage of variable nucleotides sites, nucleotide diversity, number of alleles, and pairwise ratios of nonsynonymous substitutions to synonymous substitutions (dN/dS). The r/m parameter, calculated using ClonalFrame (Didelot and Falush, 2007), is the ratio of probabilities that a given site is altered through recombination and mutation. Recombination events were tested using RDP3.44 (Martin et al., 2010). MEGA v4.1 (Tamura et al., 2007) was used to construct the minimum-evolution (ME) tree of concatenated sequences of 112 STs (including 52 STs unique from this study out of a total of 61, 9 STs from this study that shared the STs from other countries in the database, and 51 STs from different continents that formed CCs or groups as analyzed by eBURST) (Supplementary Table S2). Statistical confidence of the nodes in the ME tree was assessed by bootstrap interior branch test (500 replicates).
Results
Allelic profiles
Of the 71 isolates, there were 61 STs (Table 1). Nearly 10% of the isolates belonged to ST3, all being clinical isolates collected from 2002 to 2008. Three environmental/seafood isolates (ZS99, MJ3, and C5-1) from samples in Zhoushan (north coast of Zhejiang province) fell into ST163, and two isolates from shrimps fell into ST208. ST120 included two clinical strains (HM4 and HM17). The remaining 57 STs were represented by 57 isolates, including 8 clinical strains, 1 reference strain BJ1997, and 48 seafood-related isolates. We did not find clonally related STs between clinical strains and the seafood-related isolates that tested positive for pathogenic markers from our strain collection. However, there were clonal relationships with ST3, ST120, and ST189 between clinical strains from this study and those from environmental sources in other parts of China (Supplementary Table S2).
Analysis of CCs, groups, and singletons
The eBURST analysis divided 201 STs into 4 CCs, 18 groups, and 141 singletons (Supplementary Fig. S1). Two STs from this study (ST3, ST161) were contained in two CCs (CC3 and CC49), whereas seven other STs (ST1, ST189, ST120, ST203, ST168, ST184, and ST153) were in six groups (groups 1–6) (Supplementary Table S2). ST3 covered seven clinical isolates from this study (41%, 7/17; Table 1) and belonged to the ancestral CC3, which were joined by ST2, ST27, ST42, ST51, ST71, ST72, and ST192 (Supplementary Fig. S1). Of 60 STs containing 206 strains, there were only 15 STs that all contained clinical strains (n=131 strains), mostly in CC3 (109 strains, 83.2%) or its seven SLVs (7 STs, n=7), and groups 1 (3 STs, n=10), 4 (2 STs, n=3), and 15 (2 STs, n=2). The STs in other groups or CCs were either from environmental sources or a mixture of clinical and environmental sources (Supplementary Table S2). The clinical strains in the CC3 or its SLVs were from 12 countries including China, covering a 14-year span from 1996 to 2009. Multiple serotypes were found in CC3 (O1:K25, O1:KUT, O3:K6, O3:K68, O3:Kuk, O4:K8, O4:K68, O5:K68). CC49 was comprised of four STs (ST49, ST53, ST70, and ST161), represented by four environmental isolates from United States, Norway, Chile, and China (Supplementary Table S2), with ST49 being the founding genotype (Supplementary Fig. S1). The ST211 isolate was from clam with O3:K6 serotype and appeared to be a DLV of ST3 for its differences in the loci recA and dtdS (Table 1).
Phylogenetic analysis
Figure 1 shows that the 112 STs formed four branches. The vast majority of the strains from our laboratory were in the two main branches together with those from other parts of China or other countries. There were six strains (belonging to five different STs) branched off as two different clusters: ST207, ST209, and ST211 as one cluster, and ST208 and ST210 as the other. The recA gene from these strains was diversified (80%–82% nucleotide identity) from typical V. parahaemolyticus strains, but had 82%–84% identity to V. fortis (AJ842423), V. tubiashii (AJ842522), and V. halioticoli (AJ842431) (Table 2). Cluster analysis by recA alone gave the same structure (Fig. 1, insert), indicating that outlining of these strains was mainly due to recA diversification, most probably by recombination as shown by an r/m ratio of 3.038 (Table 3). The recA locus of the 71 isolates tested did show six unique events of recombination as detected by the RDP program.
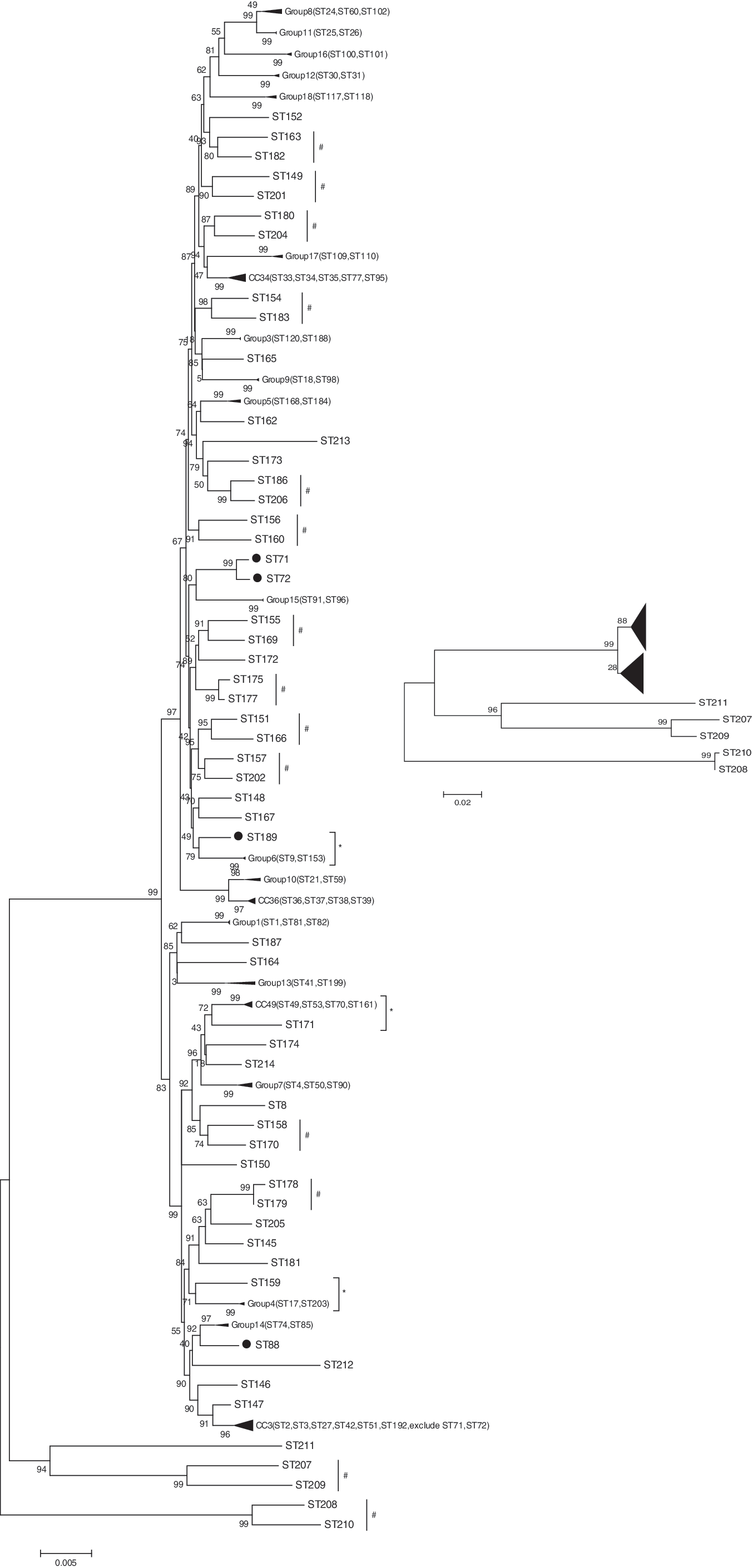
The minimum evolution tree of 112 STs based on concatenated sequences (representing all STs from this study, and those from different continents that formed clonal complexes or groups as shown by eBURST analysis; Supplementary Table S2). The inset is the tree of recA loci from the 112 STs, showing that the divergence of the recA loci in these five STs from this study contributed to the branching of the major tree. #Close relationship between two STs; *close relationship between STs in this study and those of clonal complexes or groups; •, STs in minimum-evolution tree divergent from eBURST results; ◂, the symbol of varying size means that those STs are clustered in the same branch as opposed to the neighboring one and is used to save spaces for other STs with more pronounced divergence.
ST, sequence type.
Based on all 71 isolates tested in this study. The ratio of probabilities that a given site is altered through recombination and mutation represents a measure of how important the effect of recombination is in the diversification of the sample relative to mutation.
recA recombination events using RDP3.44 (Martin et al., 2010).
The tree revealed two major differences with eBURST analysis (Supplementary Fig. S1 vs. Fig. 1). ST71 and ST72 that belonged to the same CC3 were clustered in the ME tree with ST91 and ST96 of group 15, which was far from CC3 in eBURST. ST88 and ST189, though being in the same group 2 by eBURST, were in different clusters corresponding to group 14 (ST74 and ST85) and group 6 (ST9 and ST153), respectively, in the tree. ST211 (strain KP34) was DLV of CC3, which was placed in the outer branch because of its divergent recA sequence (Fig. 1, inset).
Nucleotide diversity and recombination
The number of polymorphic sites of the loci tested were from 28 (pyrC, tnaA) to 255 (recA) and the number of alleles ranged from 32 (tnaA) and 52 (gyrB) in the seven loci of 71 V. parahaemolyticus isolates (Table 3). The percentage of variable sites and nucleotide diversity per site were greatest in recA (34.9% and 0.0564, respectively), followed by dtdS (15.1% and 0.0278). The other loci had variable sites of 5.7%–9.1%. The low ratio of dN/dS suggests that negative selection was dominant in these house-keeping genes. Of the seven loci, six had the average per site r/m ratio between 0.67 and 1.23, indicating that recombination and mutation did not differ significantly on diversification of these loci. With recA, however, recombination appeared to play a much greater role than mutation in the generation of genetic heterogeneity in the strains tested, as evidenced by its average r/m ratio of 3.038 (Table 3).
Discussion
One of the major findings in this study was that seven clinical isolates belonging to ST3 linked with the pandemic CC3 (Supplementary Fig. S1) were also existent in China as early as in the year 2002, a year earlier than we previously reported (Vongxay et al., 2008) and as recently reported by Yan et al. (2011). However, it does not exclude the possibility that such strains could have emerged in China before 2002, as was the case in Japan and Taiwan in 1996 (Okuda et al., 1997; Chiou et al., 2000). The CC3 strains, represented by pandemic O3:K6 serotype, were first found in India in 1996 and then in other Asian countries, the American continent, Europe, and even Africa (Martinez-Urtaza et al., 2005; Nair et al., 2007; Ansaruzzaman et al., 2008). However, in this study, no clonally related STs were identified between the clinical strains and seafood-related isolates that were TDH positive (Table 1). The number of thermostable direct hemolysin (TDH)-positive strains in this study seems to be low (about 8% from our strain collection). The low proportion of TDH-positive strains in our collection could be due to the difficulty in isolating TDH-producing colonies on agar plates as a result of relatively low proportion of TDH-producing V. parahaemolyticus to total V. parahaemolyticus in some samples (DePaola et al., 2000). However, there were clonal relationships with ST3, ST120, and ST189 between clinical strains from this study and those from environmental sources in other parts of China (Supplementary Table S2).
Nair et al. (2007) have indicated that there are currently 21 serotypes reported as having the same genetic lineage of pandemic O3:K6 as shown by identical ribotyping or PFGE profiles. A diverse set of serotypes belong to the same genetic lineage of ST3 in CC3 (Supplementary Table S2), suggesting that there might be horizontal transfer of genes associated with O or K antigen synthesis among V. parahaemolyticus strains in the environments. This was proposed as the mechanism of serotype diversification of V. cholerae with identical recA ST (Colin et al., 2000). Although the average per site r/m ratios were low (0.67–1.23) on six of the seven loci examined, diversification of the recA gene was three times more likely because of recombination than mutation (Table 3). RDP analysis on the recA loci of 71 isolates showed six unique recombination events, whereas there were none for the other six loci. This result seems to be contradictory to earlier studies that showed that recombination was more likely than mutation in genetic diversification of V. parahaemolyticus (Gonzalez-Escalona et al., 2008; Yan et al., 2011). Prediction results may vary with different software using different algorithms
Of the four branches in the phylogenetic tree, the vast majority were in the two major branches, in which the STs from this study were interspersed with those from other countries, indicating that these Chinese strains do not cluster by geographic origins but are as diverse as those from other countries. There were five STs in this study that fell off the major branches (Fig. 1). This divergence was largely attributable to high numbers of polymorphic sites (Table 3) in recA, as this locus alone gave the same phylogenetic structure as the branch in the major tree (Fig. 1 insert). The recA genes from strains of these STs were divergent from “typical” V. parahaemolyticus strains (Table 3), but had 82%–84% identity to V. fortis, V. tubiashii, or V. halioticoli and, more strikingly, 73.3%–76.9% identity to the CDS24 of a Vibrio sp. plasmid p23023 (GenBank Accession No. CP000755.1) (Hazen et al., 2007). These results appear to indicate that the recA gene could have been recombined by lateral gene transfer and/or gene conversion. Such possibility was suggested with the 16S rRNA genes of different strains of V. parahaemolyticus (González-Escalona et al., 2005).
In conclusion, the present study reveals emergence of pandemic CC3 strains as early as in the year 2002 in China. The recA gene of some Chinese V. parahaemolyticus isolates was found divergent from a majority of the strains examined in this study most probably by recombination. Application of the MLST scheme to more V. parahaemolyticus strains and by different laboratories would facilitate a global picture of the epidemiology and genetic population structure of this seafood-borne pathogen.
Footnotes
Acknowledgments
This research was funded in part by the Key Project of National Science and Technology Pillar Program (2009BADB9B01) and the Natural Science Foundation of China (30571436 to W.F. and 30700605 to B.W.). The authors thank Dr. N. Gonzalez-Escalona at the Center for Food Safety and Applied Nutrition, Food and Drug Administration, for communicating with the sequence verification. The authors appreciate Dr. Lingling MEI at Zhejiang Provincial Center for Disease Control, Hangzhou, Zhejiang, for kindly providing the clinical strains.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.