
Abstract
The European Union Reference Laboratory for Listeria monocytogenes (EURL for L. monocytogenes) coordinates a European network of 29 National Reference Laboratories (NRLs). Depending on a national decision, NRLs undertake food, environmental, and veterinary L. monocytogenes strain surveillance in their respective countries. In the framework of the PulseNet Europe network, two pulsed-field gel electrophoresis (PFGE) subtyping proficiency testing (PT) trials were carried out in 2003 and 2006. The obtained data showed that PFGE profiles can be compared and exchanged between laboratories. However, no further PT trial had been performed since 2006. In this context, two PT trials were organized by the EURL to evaluate the ability of NRLs to perform conventional serotyping, molecular serotyping and PFGE subtyping. Eleven well-characterized isolates of L. monocytogenes were used: six and nine isolates were tested in 2009 and 2010, respectively. Three isolates were repeated between the two studies. In the 2010 panel, a strain was tested in duplicate, and two strains were related to the same epidemiological group. The strains were analyzed blind in different laboratories (17 in 2009 and 25 in 2010) using (1) their own in-house method for serotyping methods and (2) standardized protocols based on the PulseNet protocol for PFGE. For conventional serotyping, 86.0% in 2009 and 91.0% in 2010 of the serotypes obtained were in agreement with the EURL data. For molecular serotyping, 93.5% of the results in 2009 and 95.2% in 2010 matched the EURL data. For PFGE, 68.9% in 2009 and 81.7% of the combined AscI/ApaI profiles were indistinguishable from the EURL reference profiles. The variations observed could be attributed to slight standardization defaults or, in a few cases, to a failure in DNA extraction. These PT trials provided a valuable opportunity to improve the subtyping ability of NRLs and facilitate exchanges of subtyping data in the future.
Introduction
Conventional serotyping by agglutination is the universally accepted method for distinguishing Listeria monocytogenes strains. Serotyping enables the identification of 13 different serotypes. Among the different methods of molecular serotyping, the method developed by Doumith et al. (2004) is the most commonly used and considers five distinct serogroups. The European Union Reference Laboratory (EURL) for L. monocytogenes has recently developed a test using Doumith's method with an additional polymerase chain reaction (PCR) based on the amplification of the flaA gene, in order to recognize atypical strains such as the EGDe strain and to assign them to the expected serogroup (Kerouanton et al., 2009). Among the available molecular subtyping methods for L. monocytogenes, pulsed-field gel electrophoresis (PFGE) has been widely applied to characterize human and food isolates for two decades (Carriere et al., 1991; Buchrieser et al., 1993; Brosch et al., 1996; Kerouanton et al., 1998).
Since 1996, the U.S. PulseNet network has used PFGE to identify clusters of L. monocytogenes among the different U.S. states (Swaminathan et al., 2001). Now, similar networks are working worldwide via PulseNet International. This system aims to strengthen subtyping capacities and to improve the surveillance of L. monocytogenes in each country. In Europe, in the framework of the PulseNet Europe network, two PFGE subtyping proficiency testing (PT) trials were carried out in 2003 and 2006 (Martin et al., 2006; Brisabois, personal communication). PulseNet Europe ceased to be active in November 2006 (Swaminathan et al., 2006). That is why, to our knowledge, no subtyping PT trial has been performed in Europe since 2006.
Out of 29 European National Reference Laboratories (NRLs) responsible for L. monocytogenes surveillance in food, only five took part in the previous PulseNet Europe PT trials. For the 24 remaining food NRLs, no information was previously available on their proficiency for subtyping L. monocytogenes. Thus, it is necessary to assess the ability of these NRLs to perform subtyping.
The network of food NRLs has been coordinated since 2006 by the EURL, located at the ANSES Maisons-Alfort Laboratory for Food Safety (
Methods
Bacterial strains
Consecutively in 2009 and 2010, the three methods were evaluated against the same strain panel. The panel used in 2009 consisted of six food strains, and the 2010 panel consisted of six food strains and three clinical strains (Table 1). Three strains were common to the two panels. In the 2010 panel, the strain 06CEB476LM was tested in duplicate, and the strains TS21 and TS55 were related to the same epidemiological group (Bille and Rocourt, 1996). All the strains referenced with a CEB collection number were food strains. The strains labeled TS (“test study”) were previously used in the study of Bille and Rocourt (1996) (Table 1). The two panels consisted of strains from the major serotypes and serogroups (Table 1). The strain TS02, previously serotyped as 1/2c, displayed no flagellar phase as already observed for this strain (Schönberg et al., 1996). The two 1/2a serotype strains, 05CEB504LM and 08CEB155LM, were considered as “atypical,” as observed for the EGDe reference strain (Doumith et al., 2004): Imo1118 gene amplification was positive, and flaA gene amplification was also positive. The two panels were divided into nine distinct combined PFGE profiles (Fig. 1).

Similarity dendrogram and raw image for nine distinct combined pulsed-field gel electrophoresis (PFGE) AscI/ApaI profiles of strains selected for the 2009–2010 proficiency testing trial period (Unweighted Pair Group Method with Arithmetic Mean dendrograms using Dice coefficient, with a 1% tolerance limit and 1% optimization).
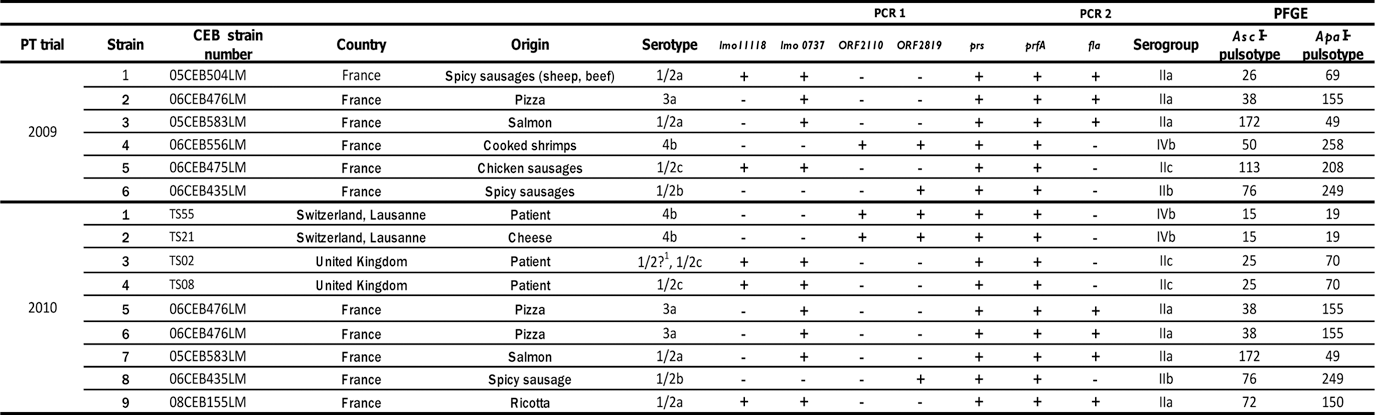
1/2? means that the strain TS02, previously typed as 1/2c by Bille and Rocourt (1996), displayed no flagellar phase in this experiment. “+” means detection of the amplified fragment of the expected size. “-“ means no detection of the amplified fragment at the expected size.
Intra-laboratory reproducibility of the subtyping methods
The duplicated strain and the three strains included in both panels were added to assess the reproducibility of each of the three methods within the same laboratory.
Discriminatory power of the subtyping methods
The discriminatory power of each method was assessed by its capacity to recognize the homogeneity of the two strains, TS21 and TS55, within the same epidemiological group.
Subtyping protocols used by EURL
For conventional serotyping, the species identification of L. monocytogenes was performed using ALOA plates (AES, Combourg, France) and CAMP Test (McKellar, 1994). Each strain was serotyped by agglutination using commercially available antisera (Denka, Eurobio, Les Ulis, France), after adapting the manufacturer's instructions and using the procedures outlined by Seeliger and Höhne (1979). The laboratory has been accredited by the French Accreditation Committee (COFRAC) for this serotyping method as an internal method (accreditation no. 1-2246, Section Laboratories;
The molecular serotyping was performed according to Kerouanton et al. (2009).
PFGE was performed using the standard U.S. Centers for Disease Control and Prevention PulseNet protocol (Halpin et al., 2010) with minor modifications: in the PulseNet USA (PN USA) extraction protocol, cell density per plug is lower than in the EURL protocol (0.9–1.0 OD 610 PN USA versus 1.6–1.8 OD 600 EURL), so the concentrations of proteinase K and other reagents were changed in the same proportion as the cell density (lysozyme, proteinase K, sodium dodecyl sulfate, and lysis buffer). The lysozyme was prepared in a TE buffer (PN USA) instead of sterile water (EURL). Lysozyme was incubated at 56°C (PN USA) instead of 37°C (EURL). The amount of restriction enzyme was higher in the PN USA protocol than in the EURL's: for AscI, 0.125 U/μL (PN USA) versus 0.100 U/μL (EURL); and for ApaI, 0.250 U/μL (PN USA) versus 0.100 U/μL (EURL). The laboratory has been accredited by the French Accreditation Committee (COFRAC) for this PFGE method as an internal method (accreditation no. 1-2246, Section Laboratories;
Profiles were analyzed and interpreted with BioNumerics software (V 5.01, Applied Maths, Kortrijk, Belgium), according to the standard operating procedure (SOP) developed at the EURL (Felix et al., 2012). It is based on the interpretation method developed by Barrett et al. (2006) and the PulseNet USA PFGE profile interpretation SOP, wherein a difference of one band is the limit to consider two PFGE profiles as different. Two profiles without observable difference (complete matching of the observed bands) are reported as indistinguishable. A down limit for band interpretation at 33 kbp was established according to EURL experience and the results of the PFGE PT trial performed on Salmonella by Peters et al. (2003).
For this study, a similarity value of 97% was established as a cut-off to consider two profiles as indistinguishable in a Unweighted Pair Group Method with Arithmetic Mean (UPGMA) dendrogram using Dice coefficient, with tolerance and optimization parameters set at 1% as advised by PulseNet Europe (Martin et al., 2006). The limit of 97% was chosen as the minimal similarity value observed in the EURL database between two indistinguishable profiles.
Interpretation of results by EURL
For serotyping, the result was considered as good if the tested strain serotype/serogroup was in agreement with the serotype/serogroup obtained by the EURL. For the TS02 strain, the somatic agglutination result was accepted despite the absence of a flagellar agglutination result.
For PFGE, the profiles analysis was performed following the criteria developed by the EURL (Felix et al., 2012) and all criteria were not taken as mandatory for this study. Some criteria had already been used by Martin et al. (2006). The profiles were analyzed independently from each other and then compared to the profiles obtained by the EURL (reference profiles). Profiles which do not comply with interpretation criteria were not analyzed and considered as having a 0% match with the reference profile. The results obtained were classified into three categories: (i) an NRL's results were considered as “good” if all profiles were indistinguishable from reference profiles (similarity value superior or equal to 97.0%); (ii) an NRL's results were considered as “satisfactory” if only 1 AscI-PFGE profile or ApaI-PFGE profile had a similarity value inferior to 97.0% with reference profiles, for the whole panel; and (iii) in all other cases, an NRL's results were considered as “unsatisfactory.”
Subtyping protocols used by the NRLs
The choice of the protocol was left to the convenience of each participant. Only PFGE gel migration parameters were set by the EURL. They were the basic requirements to match with PulseNet protocol (Graves and Swaminathan, 2001). The type of TBE buffer was recommended but not imposed. The participants were also asked to place the Salmonella Braenderup H9812 reference system every six wells on the gel and to send the gel images to the EURL in the TIF file format.
Results
In 2009 and 2010, 17 and 26 NRLs participated, respectively, in the PT trials.
Conventional serotyping
In 2009, 86.0% of the results obtained were in agreement with the EURL data. Eleven NRLs out of 16 found the expected serotype. In 2010, 91.0% of the results were in agreement with the EURL data. Ten NRLs out of 20 obtained the expected serotype.
In 2009, one NRL used a kit from Beckton Dickinson, which was not suitable for performing the entire serotyping. Regarding the other NRLs (which used the antisera kit from Denka Seiken), jointly in 2009 and 2010, 46% of the deviations were related to somatic agglutination and 53% to flagellar agglutination; the antisera I, IV, VIII, C, and D were systematically responsible for these deviations.
For the duplicated strain, 17 NRLs out of 20 obtained the same serotype. Three NRLs obtained 1/2a instead of 3a at least for one of the duplicates. For the three strains tested in both years, 16 NRLs tested the same three strains in both PT trials, and 11 of them obtained the same serotype for each of these strains. Five NRLs failed to obtain the expected serotype in 2009, but successfully serotyped these strains in 2010. For the two epidemiologically related strains, out of the 20 NRLs that participated in 2010 19 NRLs obtained the same serotype and one failed. For the TS02 strain, which displayed no flagellar phase, four NRLs failed to obtain the expected serotype, and 11 did not obtain the flagellar phase.
Molecular serotyping
The results obtained with Doumith's protocol (Doumith et al., 2004) were fully satisfactory over the 2 years. For flaA gene detection, seven NRLs obtained wrong results over the 2 years.
In 2009, 93.5% of the serogroups found were in agreement with the EURL data. Ten NRLs out of 14 obtained the expected serogroups for all the tested strains. For the atypical strain 05CEB504LM, all the NRLs obtained the expected serogroup except four NRLs, which found the IIc serogroup instead of IIa.
In 2010, 95.2% of the results were in agreement with the EURL data. Sixteen NRLs out of 20 obtained the expected serogroups.
For the atypical strains 05CEB504LM and 08CEB155LM, respectively, three NRLs and two NRLs obtained serogroup IIc instead of IIa. For the strain in duplicate, all the NRLs obtained the same serogroup. Thirteen NRLs tested the same three strains over both PT trials; all of them obtained the expected serogroup, except one which got a false negative result for the flaA gene amplification, for the strain 06CEB476LM in 2009. This NRL found the expected serotype in 2010. For the two epidemiologically related strains, all the NRLs obtained the expected serogroup, except one which obtained a false negative result for the flaA gene amplification.
PFGE gel quality analysis
For two of the 15 NRLs that attempted PFGE in 2009, the AscI-PFGE gels were not accepted at the visual step evaluation: many extra bands were observed, and parts of the bands were smeared or not visible. In 2010, one of these two NRLs again obtained unsatisfactory results and the other one a good result. In 2010, for all 17 NRLs, the gels were accepted at the visual evaluation step. In 2009 and 2010, all the ApaI-PFGE gels were accepted at the visual evaluation step.
PFGE intra-laboratory reproducibility
In 2010, for the 06CEB476LM duplicates, one NRL obtained a difference of one band between the duplicate strains (Table 2, laboratory 13), and two NRLs obtained indistinguishable combined profiles for these strains (data not shown). However the combined profiles were different from the EURL reference (Table 2, laboratories 3 and 15).
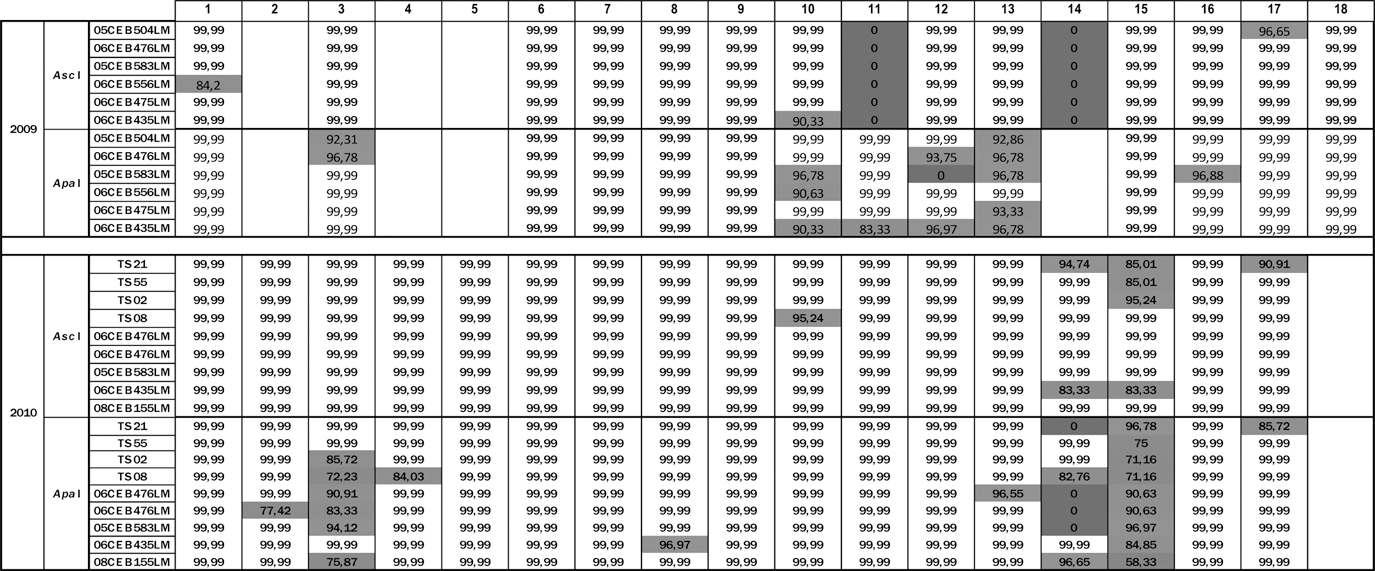
AscI and ApaI similarity values are calculated from an Unweighted Pair Group Method with Arithmetic Mean dendrogram using Dice coefficient, with tolerance and optimization set at 1%. NRLs are designated through a blind codification. Cells in white designate the results in agreement with EURL reference profiles. Cells in light gray designate failed results. Cells in dark gray designate poor quality profiles not analyzed and considered as matching at 0%. Empty cells indicate that laboratories did not participate in the proficiency testing trials for the year concerned.
For the two epidemiologically related strains, TS21 and TS55, 14 NRLs out of 17 obtained the expected combined profiles. For the TS02 and TS08 strains, 13 NRLs out of 17 obtained the expected combined profiles.
Fourteen NRLs tested the three same strains over the two PT trials. For five NRLs, the combined profiles were indistinguishable from the EURL references in 2009 and 2010 (Table 2, laboratories 1, 6, 7, 9, and 17). For four NRLs, the results improved (Table 2, laboratories 10, 11, 12 and 16) between the two PT trials. For two NRLs, the combined profiles were indistinguishable from the EURL references in 2009, but not in 2010 (Table 2, laboratories 8 and 15). For the three remaining NRLs, the combined profiles were different from the EURL references in both years (Table 2, laboratories 3, 13, and 14).
PFGE results on the total strain panel
In 2009, 68.9% of the combined profiles were indistinguishable from the EURL reference profiles. At the end of the 2009 PT trial, eight NRLs could be considered to be competent for PFGE.
In 2010, 81.7% of the combined profiles were indistinguishable from the EURL reference profiles. At the end of the 2010 PT trial, 14 NRLs could be considered as competent for PFGE.
Discussion
Seventeen (2009) and 25 (2010) NRLs participated in the two PT trials. These PT trials aimed to assess the NRLs' ability to perform three different L. monocytogenes subtyping methods: conventional serotyping, molecular serotyping, and PFGE subtyping.
For conventional serotyping, the deviations were caused either by the use of an incomplete antisera kit, inappropriate culture of the strains, poor performance of agglutination, incorrect interpretation of the results, or a fault in serum agglutination. The laboratories that used an incomplete antisera kit in 2009 ordered the whole antisera kit immediately after the PT trial and obtained satisfactory results for the 2010 PT trial. In 2010, all participants used the Denka Seiken Listeria monocytogenes specific agglutination kit. The sera responsible for abnormal or weak agglutinations were often reported in the past in our laboratory as faulty and related to poor serum quality. The same conclusions were drawn in the study of Schönberg et al. (1996). Like the EURL method, the protocol used by Seeliger and Höhne took 4 days to perform. The method recommended by Denka Seiken took at least 4 days to perform, depending on the growth potential of the strain. The results obtained here did not indicate that any one protocol was better than the others.
For molecular serotyping, the results obtained with the protocol of Doumith et al. (2004) were fully satisfactory in both years, in accordance with the conclusion drawn from the multicenter test organized by Doumith et al. (2005). We may consider that molecular serotyping according to Doumith et al. (2004) was satisfactorily performed by the NRLs. The seven false negative or positive results obtained by NRLs over the 2 years were caused by wrong result interpretation or an inappropriate implementation of the flaA PCR protocol.
For PFGE, the main deviations observed were the presence of supplementary bands or the lack of bands in the profile. These deviations were likely related to incomplete restriction, mostly due to a poor quality of the DNA, as previously shown by Martin et al. (2006). Contamination of the reagents, buffers, and purified water used during the extraction are primarily suspected. However, in a few cases, the appearance or disappearance of bands was observed even for good quality profiles and remain unexplained. In this study, the unexplained deviations represented 1% of the deviations observed in 2009 and 2010. The unexplained deviation could be reduced by improving the extraction efficiency. For example, as mentioned in Halpin et al. (2010), lysozyme was much more active at 56°C than at 37°C providing a more reliable complete lysis of the cells. This parameter should be recommended in the future to the participants as an extraction improvement basis.
The second cause of deviation was distortion of DNA migration, resulting in a shifting of profile compared to the EURL reference. These distortions were due to inappropriate migration parameters, i.e., buffer composition, buffer volume, gel quality/concentration, electrophoresis cell level, gel casting stand level, voltage/amperage regulation, switch time or switch time ramping regulation, electrode statement, and temperature regulation. Slightly distorted profiles were also observed for three NRLs. One of these NRLs used all the migration parameters recommended by the EURL and also used the same buffer reference. For this NRL, the migration system was questioned. Moreover, when implementing the same protocol, distortions disappeared when using another similar PFGE system. We confirmed here that the migration system can be one of the causes of slight migration distortion. According to the supplier of the migration system, minor distortion could be related to the temperature stability in the migration chamber or a malfunction of the generator, as noted by Murchan et al. (2003). It is thus necessary to focus on these two parameters in the development of a PFGE protocol, by monitoring the buffer temperature throughout the run, as a quality control parameter for example. The buffer type and buffer volume used could also explain slight distortions. However, five different buffer types were used among all the participants, and no correlation between the buffer type and distortion was found.
The third cause of deviation was band separation failure resulting in profiles with abnormal band shapes. Different parameters such as migration conditions (abnormal variation of the buffer temperature) and the manner in which DNA plug slices were inserted into the gel could explain these deviations. This could be solved easily by improving technical practices.
Coloration failures, generating over-exposed or excessively weak signals of the profile, were also observed. These deviations could easily be solved by adjusting the size of the DNA plug (which could be cut smaller) and/or by adjusting exposure time to ethidium bromide revelation.
Most of the problems encountered by the participants could easily be solved. Over the 2 years, the NRLs improved their results or remained stable, except for two NRLs that showed a significant decrease in their result quality. This could be explained by changes in their internal organization. NRLs that obtained unsatisfactory results for these PT trials were invited to repeat their analysis after implementing corrective measures.
Conclusion
The NRLs that participated successfully in the PT trials are considered to be competent for conventional and molecular serotyping of L. monocytogenes, as well as PFGE subtyping. These PT trials helped to strengthen the NRLs' subtyping capacity and to harmonize the subtyping of L. monocytogenes at European Union level.
It is expected that these 4 years of efforts to standardize subtyping methods, associated with training of the NRL network, as well as with assessment and improvement of NRL competence, will be a good basis for a European PFGE database for L. monocytogenes isolated from food, environment and animals, whose creation is currently being considered.
Footnotes
Acknowledgments
We thank the study group of European NRLs involved in the two studies. We also thank Peter Gerner-Smidt (CDC) for his helpful advises in the writing of this publication. This work was conducted as part of the activities of the European Union Reference Laboratory for Listeria monocytogenes and was supported by a grant from the Directorate-General for Heath and Consumers (DG Sanco) of the European Commission.
Disclosure Statement
No competing financial interests exist.