
Abstract
Staphylococcal enterotoxin D and R (SED, SER) production was determined in 24 S. aureus strains harboring sed gene. Seven of them were not able to produce SED as evidenced by enzyme-linked immunosorbent assay and Western blotting. Sequencing revealed that all these strains harbor a variant of sed gene. Expression of SER was detectable in 22 out of 24 isolates, with variance in productivity ranging from ∼40 to 450 ng/mL. Out of the seven isolates not able to produce SED, three produced high amounts of SER (249–396 ng/mL), two produced less than 200 ng/mL of SER, and two were found to express no detectable amount of SER. Three of those were assigned to spa type t1677 with two being of agr type III and one of agr type I. One strain was t084, agr type II, one t603, agr type II, one 2920, agr type III, one t2920, agr type III, and one t5160, agr type I. Because conventional screening procedures involve only the detection of classical enterotoxins in food, the isolates not able to produce SED presented in this study could pose a threat to human health due to SER production.
Introduction
Staphylococcal enterotoxin D (SED) was shown to be encoded, together with SER and SElJ, on the penicillinase-type pIB485-related plasmid. Together with SEA, SED is responsible for most cases of SFP (Ferry and Etienne, 2009). Staphylococcal enterotoxin R (SER) was first described by Omoe et al. (2003). It is encoded on the pIB485-related and the pF5-related plasmid. SER was shown to posses emetic activity in the monkey feeding test (Ono et al., 2008), which implies its potential involvement in SFP. The aim of this work was to study SER productivity on both the transcript and protein levels in sed-positive S. aureus strains.
Methods
Bacterial strains and growth conditions
We analyzed 24 S. aureus strains harboring the sed gene from our collection isolated previously from human intestinal tract (in total, 240 strains) (Lis et al., 2009) and food (in total, 182 strains; some of these strains were already described by Bania et al. [2006]). Enterotoxigenic reference S. aureus strains used as positive controls were kindly provided by Dr. Gerard Lina of the Centre National de Référence des Toxémies Staphylococciques, Faculté de Médecine, Lyon, France. S. aureus was grown in brain-heart infusion (BHI) broth supplemented with 1% yeast extract at 37°C, 230 rpm, overnight. Then the cultures were diluted to optical density at 600 nm (OD600) 0.1 in fresh medium and grown at 37°C, 230 rpm for 24 h. A growth curve was established for three randomly selected S. aureus strains (one isolated from food and two of human origin) using plate counts. One milliliter of culture was centrifuged for 2 min at 10,000×g, and bacterial pellets served for RNA preparation. For the Western blot and enzyme-linked immunosorbent assay (ELISA), isolates were grown in TECRA® enrichment broth (3M, Warsaw, Poland) at 37°C, 230 rpm for 24 h.
Detection of enterotoxin genes
The detection of sea-see genes was performed with the method described by Sharma et al. (2000). The selu gene was detected using the method described by Letertre et al. (2003). For the detection of tst gene, primers and conditions as described by Monday and Bohach (1999) were applied. Detection of seg, sei, selm, seln, selo, seh, selj, selk, sell, and selp genes was as previously described (Bania et al., 2006; Lis et al., 2009). Primers used for sequencing were as follows: sed-gene-for: AGGATCAAATATATTGATATAATGAAAGTG, encompassing positions 1–30; and sed-gene-rev: GAAATGGCTTTAGTGTCTGATGTTAAAG, encompassing positions 1103–1130 of complete sed gene (GenBank accession number M94872).
agr and spa typing
agr typing was performed according to Lina et al. (2003). spa typing was performed according to Harmsen et al. (2003). The sequence of the repeat-containing region of the spa gene was obtained from both strands of the polymerase chain reaction (PCR) product. Sequencing was performed using the BigDye™ Terminator Ready Reaction Cycle Sequencing kit (Genomed, Warsaw, Poland). The analysis of repeats and assignment of spa type was performed using the resources of Ridom Spa Server (
Cloning, expression, and purification of rSER
The S. aureus food isolate 214 carrying the sea, sed, selj, and ser genes was selected as a source for cloning. The region of the ser gene encoding mature SER was PCR-amplified using the AccuTaq™ LA DNA polymerase (Sigma-Aldrich, Poznań, Poland) using the protocol: 98°C (30 s), followed by 35 cycles of 94°C (20 s), 55°C (20 s), and 68°C (90 s), with a final extension at 68°C for 10 min. Cloning primers were as follows: forward: 5′- CATGCCATGGGCAAACCAGATCCAAGGCC -3′ and reverse: 5′- CATGCTGGAGCATTGTAGTCAGGTGAAG -3′; and carried the restriction sites for NcoI and XhoI, respectively. PCR product was purified from the agarose gel, digested with XhoI and NcoI (Fermentas, Vilnius, Lithuania), ligated into the pET-22b plasmid vector (Novagen, Warsaw, Poland), and introduced into DH5α Escherichia coli cells (Novagen) using heat shock at 42°C for 45s. Plasmid DNA was purified using the NucleoSpin® Plasmid (Macherey-Nagel, Düren, Germany) and sequenced. Vector containing the intact sequence of the respective region of ser was transformed into Rosetta E. coli cells (Novagen). Expression was performed according to the autoinduction protocol by Studier (2005). The purification of recombinant enterotoxin was performed on His-Select® Cobalt Affinity Gel (Sigma-Aldrich), with on-column refolding. Briefly, the bacterial pellet was lysed in 20 mM Tris-Cl (pH 8.0), containing 50 mM NaH2PO4, 150 mM NaCl, 8 M urea, and 5% glycerol. The lysate was centrifuged for 45 min at 16,000×g, and the supernatant was applied on column pre-equilibrated with the lysis buffer. The column was washed with lysis buffer, followed by washes with 20 mM Tris-Cl (pH 8.0), 50 mM NaH2PO4, 150 mM NaCl, and 5% glycerol containing decreasing concentrations of urea, from 6M to 0M. The protein was eluted with 250 mM imidazole in 20 mM Tris-Cl (pH 8.0), 50 mM NaH2PO4, 150 mM NaCl, and 5% glycerol. Protein concentration was measured using the Bradford reagent (Sigma-Aldrich). Purity of rSER preparation was checked with sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE).
Antisera preparation
Anti-SER sera were obtained by immunizing rabbits with 30 μg of rSER. Immunization was repeated four times at 3-week intervals. At 14 days after last immunization, the animals were bled and serum was prepared. The antibody titer was monitored by indirect ELISA (Omoe et al., 2003). Monospecific polyclonal anti-SER antibodies were obtained by affinity purification of sera on rSER coupled to CNBr-activated Sepharose® 4B (Sigma-Aldrich). All experiments on animals were performed in compliance with the Bioethical Committee of the Wroclaw University of Environmental and Life Sciences and guidelines for the experimentation on animals.
Western blot
The protein concentration in culture supernatants was measured using the Bradford assay (Sigma-Aldrich), and 5 μg of protein was loaded per well on SDS-PAGE. Western immunoblots were probed with antibodies against SED (Acris, Herford, Germany). Anti-rabbit-HRP (Sigma-Aldrich) was used as a secondary antibody. The blots were developed using the ECL Lumi-LightPLUS substrate (Roche, Warsaw, Poland), according to manufacturer's instructions.
Sandwich ELISA
The detection of SED was carried out using 3M™ Tecra™ Staph Enterotoxin ID Test, according to the manufacturer's instructions. SER was detected using sandwich ELISA performed according to the protocol described by Freed et al. (1982), with modifications. Briefly, culture supernatants from S. aureus strains were preincubated with 20% normal rabbit serum, in order to bind protein A, and diluted 10–20 times in phosphate-buffered saline containing 0.05% Tween-20. The antibody was biotinylated with biotin N-hydroxysuccinimide ester (Sigma-Aldrich). Conjugate of HRP-streptavidin (Sigma-Aldrich) was used to detect biotinylated antibody, and 3.3′,5.5′-tetramethylbenzidine (Sigma-Aldrich) was used as a substrate for HRP. The specificity of the ELISA was established using culture supernatants of S. aureus reference strains FRI913, A90322, FRI1151m, and CCM5757 as controls for enterotoxins SEA, SEB, SEC, SED, SEE, SEG, SEI, SElJ, SElK, SElL, SElM, SElN, SElO, SElP, and SER. rSER served as a standard in ELISA. In order to establish detection limit of SER in food, rSER was added to cow's ultra-high-temperature (UHT) processed milk at concentrations ranging from 10 pg/mL to 100 ng/mL and tested with ELISA.
RNA extraction and qRT-PCR
Bacterial pellets were suspended in 70 μL of 100 mM Tris-HCl (pH 7.4), containing 28 U/mL of lysostaphin (A&A Biotechnology, Gdańsk, Poland) and incubated for 15 min at 37°C. RNA was extracted with TRI Reagent® (Sigma-Aldrich) according to the manufacturer's instructions. RNA was dissolved in 50 μL of water and quantified by measuring A260 and A280. One microgram of RNA was treated with RNAse-free DNase I (Sigma-Aldrich) in order to eliminate residual genomic DNA. Complementary DNA (cDNA) was synthesized using random hexamers and SuperScript III® (Life Technologies, Warsaw, Poland) following manufacturer's instructions. Primers used in real-time PCR were taken from Derzelle et al. (2009). Primers for rpoB gene, selected by Derzelle et al. (2009) as one of the most stable internal control genes (Rpofor: 5′CTACAAAACCAATTCCGTATCG-3′; Rporev: 5′TTAATTGTTGAGGTGTGATAGAC-3′), were used for normalization of cDNA. Real-time quantitative PCR was carried out in iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad, Warsaw, Poland), using iQ™ Sybr® Green Supermix (Bio-Rad). The reaction mixture contained 1 μL of template cDNA, 0.5 μM of each primer, 10 μL of iQ Sybr Green Supermix, and water up to 20 μL. The reaction protocol was as follows: 95°C, 30 s; 35 repeats of 95°C, 10 s, 55°C, 15 s, and 72°C, 15 s. Specificity of PCR was evaluated by melt curve analysis in a temperature range from 90°C to 65°C performed for each reaction. Residual DNA contamination was checked in each RNA sample by running no–reverse transcriptase (no-RT) controls. PCR efficiencies for each primer pair were determined on genomic S. aureus DNA from respective reference strains by running serial fivefold dilutions of the template. Determined efficiencies were taken into account when calculating relative transcript levels according to Pfaffl (2001). Two or three independent cultures were performed for each strain.
Results
SED productivity of 24 S. aureus strains
Out of the 24 S. aureus strains harboring the sed gene, 17 strains gave a positive signal for SED in ELISA. The remaining seven did not produce SED as determined by ELISA.
Sequencing of sed gene in 24 S. aureus strains
In 24 sed-positive strains of human and food origin, the partial sed gene sequence was determined. Analysis of sed gene between positions 30 and 1103 revealed a deletion of adenosine at position 521 of the sed coding region in a motif containing eight consecutive adenosine residues in all seven S. aureus strains that produced negative result in ELISA for SED. Analysis of translated sequence of sed variant gene (sed-v) revealed that adenosine deletion should result in a premature stop codon at the position 547 of the sed coding region, corresponding to 14.7-kDa polypeptide. No mutations in ribosome binding site (RBS) between sed and sed-v genes were found.
Western blot analysis of SED production in S. aureus strains harboring sed-v gene
Extracellular proteins from the culture supernatants from the seven strains carrying sed-v gene and from reference strain F1151m with the intact sed gene were resolved in SDS-PAGE, and probed with an anti-SED antibody. SED expression could not be detected in isolates carrying the sed-v gene. None of the isolates with sed-v showed expression of the 14.7-kDa variant of SED either.
SER productivity of 24 S. aureus strains
All 24 sed-positive S. aureus strains and the reference strain F1151m were examined for SER production in the late stationary phase of growth. SER expression was detectable in 22 out of 24 isolates with variance in productivities between strains. Concentrations of SER ranged from ∼40 to 450 ng/mL. It is notable that out of the seven isolates harboring sed-v gene, three (193, 640, and 1164) produced high amounts of SER (396, 230, and 249 ng/mL, respectively), two (496, 584) produced less than 200 ng/mL of SER, and two of them (265 and 270) were found to express no detectable amount of SER (Fig. 1). We demonstrated that rSER can be detected in milk at 1.5 ng/mL concentration.
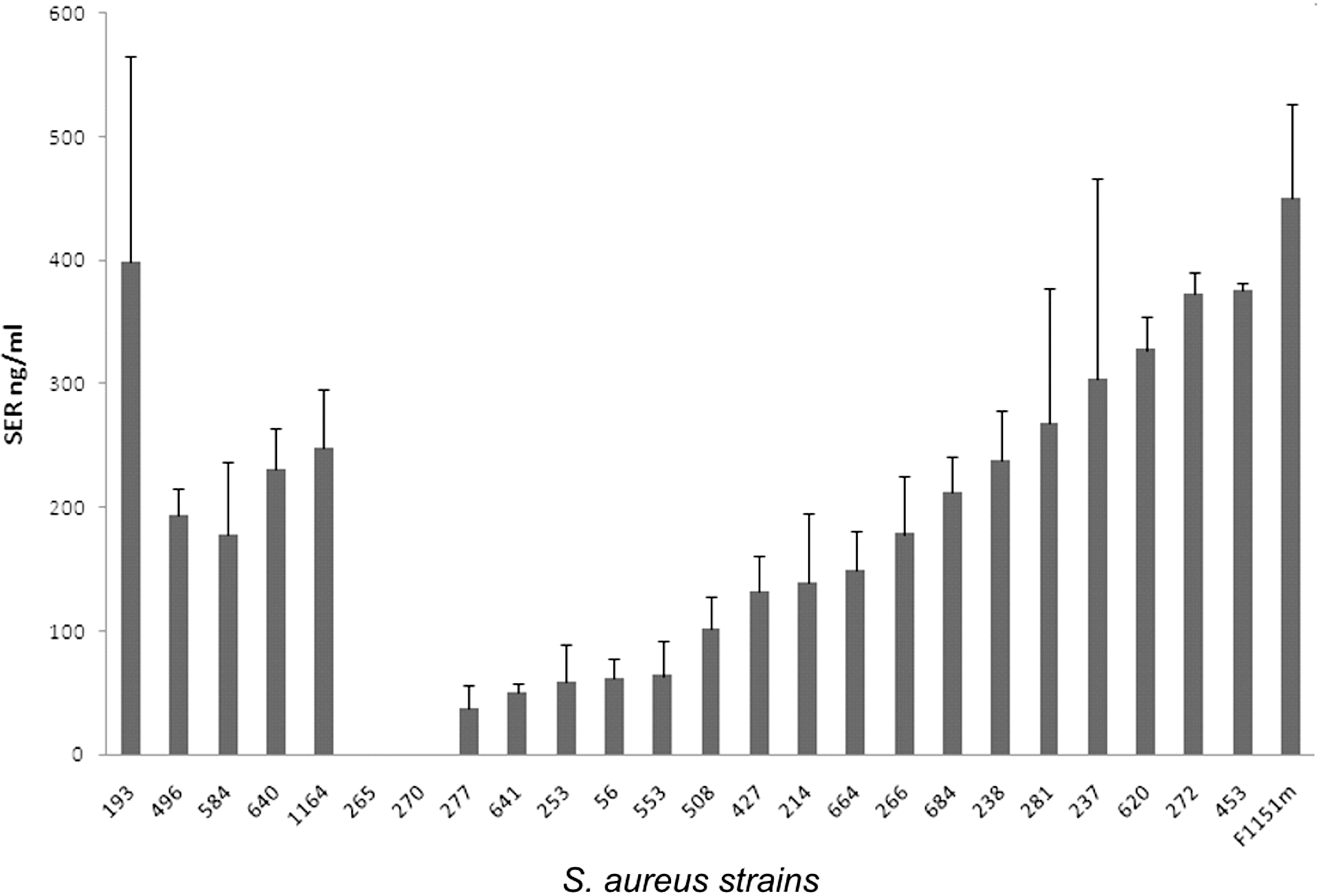
Staphylococcal enterotoxin R (SER) productivities in Staphylococcus aureus isolates in the late stationary phase of growth measured by enzyme-linked immunosorbent assay (ELISA). Staphylococcus aureus isolates 193, 496, 584, 640, 1164, 265, and 270 carried sed-v gene; remaining isolates carried intact sed. Two independent cultures were done for each strain; error bars indicate standard deviations.
qRT-PCR analysis of sed and ser genes expression
A comparison of ser and sed transcripts after 24 h of culture was done for the seven isolates with sed-v gene. In five isolates, both sed and ser transcripts were detected, whilst in two isolates (270 and 265) no sed and ser transcription could be observed (Fig. 2).
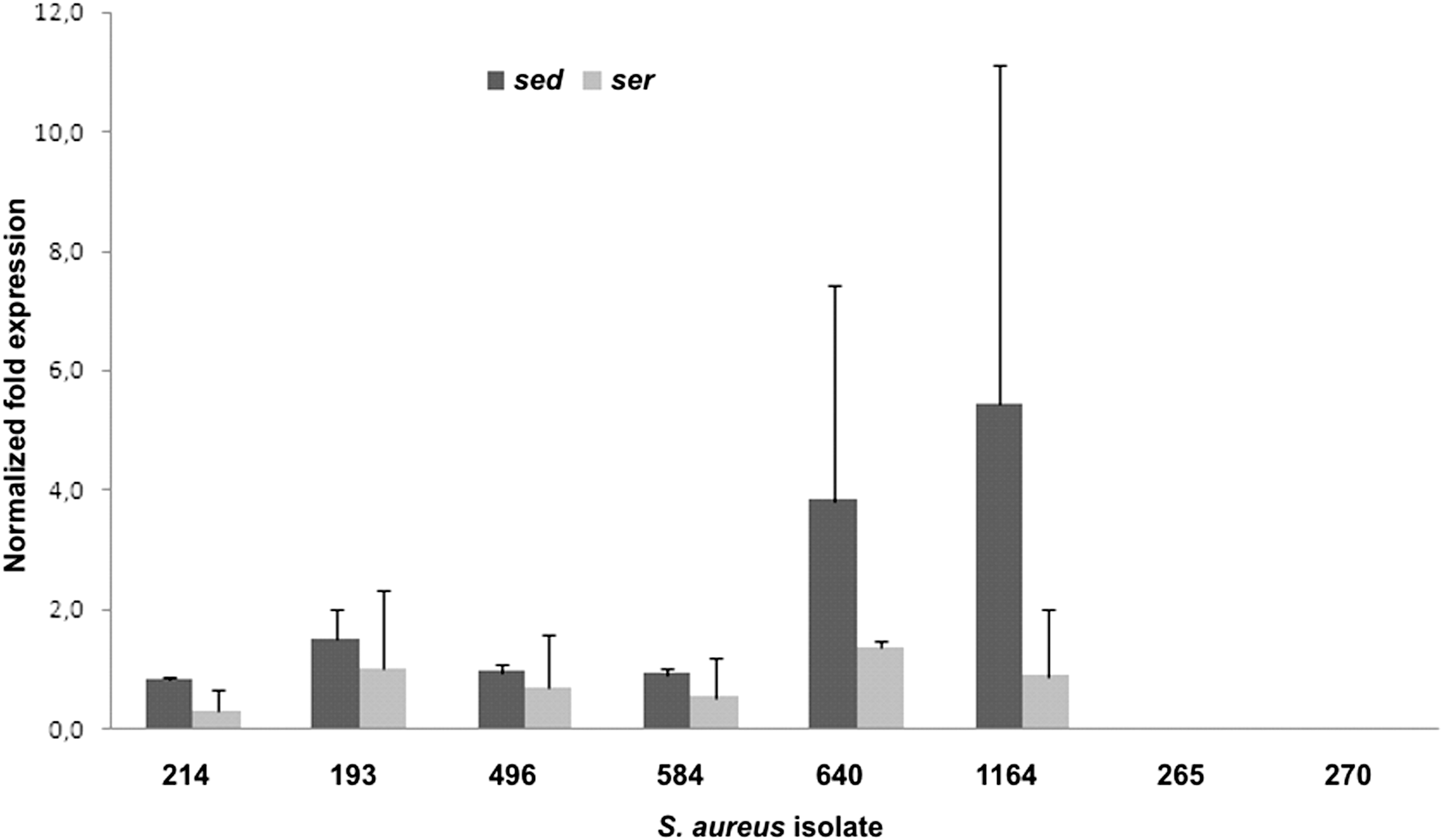
Comparison of mean relative sed and ser transcript levels in the late stationary phase of growth for the strains not able to produce Staphylococcal enterotoxin D (SED; Staphylococcus aureus isolates 193–270). The 214 isolate expressing both SED and Staphylococcal enterotoxin R (SER) was used as a positive control. Two independent cultures were done for each strain; error bars indicate standard deviations.
spa and agr types of S. aureus isolates harboring sed-v gene
Three sed-v gene–positive S. aureus strains were assigned to t1677 spa type with two of these being agr type III, and one agr type I. One strain was t084, agr type II; one t603, agr type II; one 2920, agr type III; one t2920, agr type III; and one t5160, agr type I.
Discussion
Recent studies have shown that novel SEs can be responsible for staphylococcal gastroenteritis outbreaks (Chen et al., 2004; Ikeda et al., 2005), but with limited data available it remains difficult to predict the threat they pose to public health.
So far, a significant number of characterized staphylococcal strains harbor multiple enterotoxin genes, including combinations of classical and newly characterized ones. Biological significance of this redundancy still remains unclear, but it is possible that it helps the pathogen to maintain the pool of enterotoxin in the case of expression failure of a particular SE gene.
Twenty-four S. aureus strains from food and humans, positive for both sed and ser genes, were screened for SED production using a commercially available ELISA. Seventeen strains gave positive signal for SED, and in seven strains SED could not be detected. In order to determine the cause of SED production failure we searched for differences between SED producers and non-producing strains. Sequencing the sed gene in all S. aureus strains led to the discovery of variants of sed gene in all of the seven non-producing strains. Western blotting using polyclonal αSED antiserum revealed neither expression of complete nor truncated variant of SED. SED RNA was detected in five non-producing strains on the level comparable with strains expressing SED protein, suggesting that SED expression blockade in these strains is likely not related to RNA stability and occurs at the level of protein synthesis. Jin et al. (2002) demonstrated that, depending on its context, some premature stop codons can decrease the efficiency of translation initiation even 400 bp upstream of termination signal.
Results of spa and agr typing revealed that the seven S. aureus strains containing sed-v gene belonged to five different spa and three agr alleles, suggesting that SED expression failure is neither influenced by genetic background nor by agr type.
A sandwich ELISA developed to quantify SER protein revealed that in examined strains SER concentrations ranged from ∼40 to 450 ng/mL. Notably, out of seven SED non-producing strains, five expressed SER in amounts ranging from 180 to nearly 400 ng/mL. The results presented in this article show that the routinely used PCR and ELISA for the detection and quantitation of classical SEs in food might be insufficient. Seven S. aureus strains, belonging to diverse S. aureus clones, were found negative for SED production. A significant part of them was shown to produce SER. SER is known to posses emetic properties, at concentrations over 100 ng/mL; therefore, those isolates are potentially capable of causing SFP. These findings show that developing new methods, taking into account both classical and new SEs, are necessary in order to ensure the safety of consumers in the future.
Footnotes
Acknowledgments
This work was supported by the Polish Ministry of Science and Information Society Technologies (grant N N312 215136).
Disclosure Statement
No competing financial interests exist.