
Abstract
A multiplex polymerase chain reaction (PCR) assay was developed for simultaneous detection of Escherichia coli O157:H7, Bacillus cereus, Vibrio parahaemolyticus, Salmonella spp., Listeria monocytogenes, and Staphylococcus aureus in various Korean ready-to-eat foods. The six specific primer pairs for multiplex PCR were selected based on the O157 antigen (rfbE) gene of E. coli O157:H7, the DNA gyrase subunit B (gyrB) gene of B. cereus, the toxin regulatory protein (toxR) gene of V. parahaemolyticus, the invasion protein A (invA) gene of Salmonella spp., the hemolysin (hly) gene of L. monocytogenes, and the thermonuclease (nuc) gene of S. aureus. The 16S rRNA gene was targeted as an internal control gene in the presence of bacterial DNA. The specificity and sensitivity assays for multiplex primer pairs were investigated by testing different strains. When this multiplex PCR assay was applied to evaluate the validity of detecting six foodborne pathogens in artificially inoculated several ready-to-eat food samples, the assay was able to specifically simultaneously detect as few as 1 colony-forming unit/mL of each pathogen after enrichment for 12 h. Their presence in naturally contaminated samples also indicates that the developed multiplex PCR assay is an effective and informative supplement for practical use.
Introduction
A
Rapid and sensitive assays of high specificity are needed for the detection of pathogenic organisms in various foods. Recently, with the development of molecular techniques, polymerase chain reaction (PCR) has become an important tool for detecting and identifying pathogenic organisms in various foods (Sharma et al., 1999; Ramesh et al., 2002; Cabrera-García et al., 2004; Jofré et al., 2005; Park et al., 2007). The development of PCR-based methods, including multiplex PCR, have greatly improved the sensitivity, specificity, and speed of detecting pathogenic organisms (Xu et al., 2012). Lately, real-time PCR has successfully been performed to identify pathogens in various food products. However, these methods are not only very expensive for routine use in common testing laboratories, but also are limited to two or three different types of pathogenic bacteria per detection assay (Wang et al., 2007; Suo et al., 2010). In fact, the real-time PCR approach is still uncommon. Multiplex PCR assay is able to simultaneously amplify multiple gene targets by using several sets of target specific or degenerated primers in a single tube (Fratamico et al., 2000). Moreover, some of these techniques have been accepted as standard methods for testing the microbial contamination of food (Germini et al., 2009). Multiplex PCR assay, in comparison with uniplex PCR assays, could save considerable time and workload, and improve efficiency (Jofré et al., 2005; Park et al., 2006; Kim et al., 2007; Lei et al., 2008; Germini et al., 2009).
In the present study, we developed a multiplex PCR assay, combined with a 12-h incubation time, for the sensitive and reliable detection of Salmonella spp., B. cereus, V. parahaemolyticus, E. coli O157:H7, L. monocytogenes, and S. aureus. We also evaluated the applicability of this multiplex PCR assay in Korean ready-to-eat food (which consists of a complex food matrix) artificially inoculated with six foodborne pathogens and in naturally contaminated food samples (i.e., kimbab, sandwiches, and spicy chicken).
Materials and Methods
Bacterial strains and culture conditions
The bacterial strains were obtained from the American Type Culture Collection (ATCC; Manassas, VA), the National Culture Collection for Pathogens (NCCP; Seoul, Korea), and the Korean Collection for type culture (Daejeon, Korea) (Table 1). All bacterial strains were grown overnight at 37°C with continuous agitation in tryptic soy broth (TSB; Oxoid, Basingstoke, UK). The simultaneous enrichment broth (SEB), which is the original formulation of Kobayashi et al. (2009), was used for the simultaneous enrichment of E. coli O157:H7, B. cereus, V. parahaemolyticus, Salmonella spp., L. monocytogenes, and S. aureus. After incubation for 8 or 12 h at 37°C, culture broth was used for DNA extraction and subjected to the multiplex PCR assay.
ATCC, American Type Culture Collection; NCCP, National Culture Collection for Pathogens; KCTC, Korean Collection for type culture.
The viable cell counts of each bacterium were assessed by a plating method using selective media for respective bacteria. The cultures of each bacterium were serially diluted with sterile PBS, and 100-μL dilutions were spread over XLD agar (Merck, Darmstadt, Germany) for Salmonella spp., MYP agar (Oxoid) for B. cereus, TCBS agar (Difco, Becton Dickinson, Sparks, MD) for V. parahaemolyticus, sorbitol MacConkey agar (Oxoid) for E. coli O157:H7, Oxford agar (Oxoid) for L. monocytogenes, and Baird Parker agar (Difco) for S. aureus. The plates were incubated at 37°C for 24–48 h in order to allow for the microbial growth.
DNA isolation
The liquid cultures (1 mL) were centrifuged at 16,000×g for 5 min. The cell pellets were resuspended in 200 μL of the enzyme solution containing 1 mg/mL achromopeptidase (Fluka, Buchs, Switzerland) and 1 mg/mL lysozyme (Sigma, St. Louis, MO) in the Tris-EDTA buffer (Kawasaki et al., 2005). After incubation for 1 h at 37°C, the solution was mixed with 300 μL of 4 M guanidine isothiocyanate solution (Fluka) containing 2% (wt/vol) of Tween 20 (Sigma). A portion (400 μL) of the supernatant was transferred to a new tube containing 400 μL of 100% isopropanol. After mixing, the mixture was centrifuged for 10 min at 16,000×g, and the resulting DNA pellet was rinsed with 75% isopropanol. The pellet was then dissolved in 100 μL of distilled water. Two microliters of the solution were used directly for the PCR.
Oligonucleotides primers and multiplex PCR assays
The oligonucleotide primers used in this study are shown in Table 2. The target genes selected for their characteristics were the rfbE (O157 antigen) gene in E. coli O157:H7 (Nagano et al., 1998), the gyrB (DNA gyrase subunit B) gene in B. cereus (Park et al., 2007), the toxR (toxin regulatory protein) gene in V. parahaemolyticus (Kim et al., 2007), the invA (invasion protein A) gene in Salmonella spp. (Germini et al., 2009), the hly (hemolysin) gene in L. monocytogenes (Park et al., 2006), and the nuc (thermonuclease) gene in S. aureus (Zhang et al., 2009), all of which have been reported in recent publications as the most specific and reliable genetic targets for the six pathogens. The 16S rRNA gene was also targeted as an internal control of the presence of amplifiable bacterial DNA. The uniplex PCR was performed using an MJ minicycler (Bio-Rad, Hercules, CA) in reaction mixtures (AccuPower® Gold Multiplex PCR PreMix; Bioneer, Daejeon, Korea) containing 10 mM Tris-HCl, 1.5 mM MgCl2, 250 μM dNTP with a final volume of 20 μL of 0.5 μM of each primer, 2 U Top DNA polymerase, and 2 μL of template DNA. The multiplex PCR conditions were the same as the uniplex PCR assay except for the concentration of primers, and the optimized concentrations of the seven primer pairs in the multiplex were 0.3 μM for 16S-F/R; 0.8 μM for hly-F/R; 1 μM for gyr-F/R, tox-F/R, rfb-F/R, and nuc-F/R; and 1.8 μM for inv-F/R.
The reaction parameters were an initial denaturation at 95°C for 5 min, followed by 35 cycles of 95°C for 50 s, 58°C for 1 min, and 72°C for 50 s, and a final 5 min of 72°C for extension. Each PCR reaction was conducted in triplicate and the amplified products were separated by electrophoresis on a 2% agarose gel at 100 V for 50 min. The gels were stained with 0.5 μg/mL ethidium bromide solution and visualized using an ultraviolet transilluminator (Gel Doc XR system, Bio-Rad).
Multiplex PCR evaluation in artificially inoculated food samples
In order to validate the multiplex PCR assay in food, the nine Korean ready-to-eat food samples (listed in Table 3) and fresh lettuce were purchased from a local supermarket. Before inoculating experiments, the food samples were checked for the presence of E. coli O157:H7, B. cereus, V. parahaemolyticus, Salmonella spp., L. monocytogenes, and S. aureus. None of these pathogens could be detected by culture in these food samples. The six foodborne pathogens (E. coli O157:H7 NCCP11091, B. cereus ATCC14579, V. parahaemolyticus ATCC17803, Salmonella spp. ATCC14028, L. monocytogenes ATCC15313, and S. aureus ATCC6538) were cultured in TSB at 37°C for 18 h prior to decimal dilution in a sterile saline solution (0.85% NaCl) in order to obtain the nine levels of inoculation (from 100 to 108 colony-forming units [CFU]/g). Twenty-five-gram portions of food samples were inoculated with 1 mL of each level of inoculation, placed in a sterile plastic bag with a lateral filter (Nasco Whirl-Pak, Fort Atkinson, WI), and 225 mL of SEB medium was added. A noninoculated sample was included as a negative control. The mixture was homogenized in a stomacher (Pulsifier, Microgen Bioproducts Ltd., UK) for 1 min and incubated at 37°C. After 8- and 12-h incubation times, a 1-mL aliquot was collected from each sample for DNA extraction.
CFU, colony-forming units.
Application of the established multiplex PCR in natural food samples
Thirty-eight samples of different ready-to-eat food (kimbab and sandwiches) were collected from local stores. These samples were kept in a cool box and analyzed on the same day as collection. Each 25-g sample was mixed with 225 mL of SEB medium and incubated at 37°C for 12 h. Then, each 1 mL of culture broth was subjected to the multiplex PCR assay and culture-based detection.
Results and Discussion
Specificity and sensitivity for selected multiplex PCR primer sets
Primer pair selection is critical in the multiplex PCR assay for the simultaneous detection of six foodborne pathogens in order to ensure specificity and sensitivity and to avoid cross-reactions. Computer simulation of the different combinations of various reported primer pairs specific for each pathogen was performed with the aid of online-available software (Kalendar et al., 2009) in order to find the best combination for developing the multiplex PCR. A combination of suitable reported primers was found for all of the targets (Park et al., 2006; Kim et al., 2007; Zhang et al., 2009), taking into account that they should have similar melting temperatures (Tm ) and a similar size of target DNA to prevent differential yields in band amplification products. The primer pairs selected in this study were rfb-F/R for amplification of a 678-bp sequence from the rfbE gene of E. coli O157:H7, gyr-F/R for amplification of a 540-bp sequence from the gyrB gene of B. cereus, tox-F/R for amplification of a 375-bp sequence from the toxR gene of V. parahaemolyticus, inv-F/R for amplification of a 278-bp sequence from the invA gene of Salmonella spp., hly-F/R for amplification of a 210-bp sequence from the hly gene of L. monocytogenes, nuc-F/R for amplification of a 164-bp sequence from the nuc gene of S. aureus, and 16S-F/R for amplification of a 475-bp sequence from the 16S rRNA gene of bacterial DNA (Table 2 and Fig. 1).
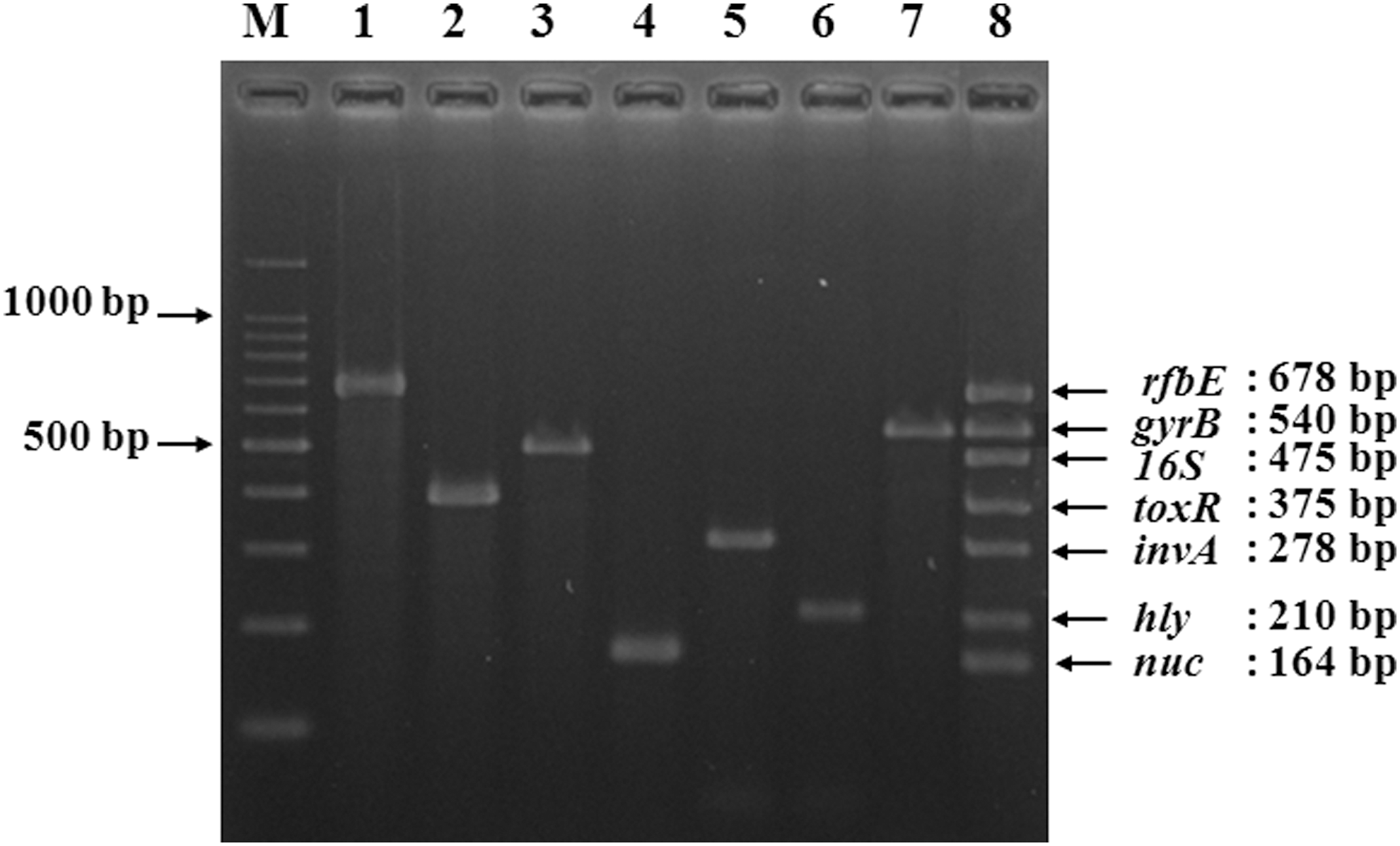
Multiplex polymerase chain reaction applied to single and multiple pathogen detection. M, 100-bp DNA ladder; lane 1, Escherichia coli O157:H7 NCCP11091; lane 2, Vibrio parahaemolyticus ATCC17803; lane 3, 16S rRNA; lane 4, Staphylococcus aureus ATCC6538; lane 5, Salmonella spp. ATCC13076; lane 6, Listeria monocytogenes ATCC19111; lane 7, Bacillus cereus ATCC14579; lane 8, the six-pathogen mixture.
With the purified genomic DNA of each pathogen, we optimized the multiplex PCR to give rise to similar amounts of each specific PCR product by adjusting the concentrations of primer pairs. Multiplex PCR was performed on the mix of DNA extracts from the six foodborne pathogens (concentration for each at 108 CFU/mL) and was compared to the same test performed on the pathogens individually (Fig. 1). The results showed that these seven primer pairs in the multiplex PCR assay worked well independently, and could distinguish the six pathogens from each other with high specificity. The findings for the optimization of annealing temperature are also shown in the image (not shown). It can be seen that lowering the annealing temperature by 2°C was required for the same loci to be co-amplified in multiplex mixtures for the multiplex PCR in the test. Based on the yield of PCR products for the seven target genes, the results showed an optimal multiplex annealing temperature of 58°C.
In order to evaluate and verify the specificity of primers in this study, each primer pair was tested by the multiplex PCR on DNA templates prepared from 18 different bacterial strains. As shown in Table 1, these seven primer pairs were specific for their corresponding target foodborne pathogens, and the nontarget pathogens also did not generate any amplification products. These results indicated that our multiplex PCR assay could be used to identify each of these six foodborne pathogens.
The sensitivity of the multiplex PCR assay was also determined by using serial dilutions (from 108 to 101 CFU/mL) of the target pathogens. DNA was extracted from overnight cultures of each pathogen grown in TSB. Although B. cereus, E. coli O157:H7, L. monocytogenes, and S. aureus could be detected down to 102 CFU/mL, simultaneous specific detection of all six pathogens could be successfully achieved down to 104 CFU/mL (Fig. 2), which is more sensitive in comparison with the detection level recognized by Kim et al. (2007) and Germini et al. (2009). In a previous study, Kim et al. (2007) assessed a multiplex PCR assay, which was able to detect at a level of 105 CFU/mL for E. coli O157:H7, Salmonella, S. aureus, L. monocytogenes, and V. parahaemolyticus grown in Luria-Bertani (LB). Germini et al. (2009) also reported that sensitivity of multiplex PCR assay for E. coli O157:H7, Salmonella, and L. monocytogenes grown overnight in TSB was 106 CFU/mL.
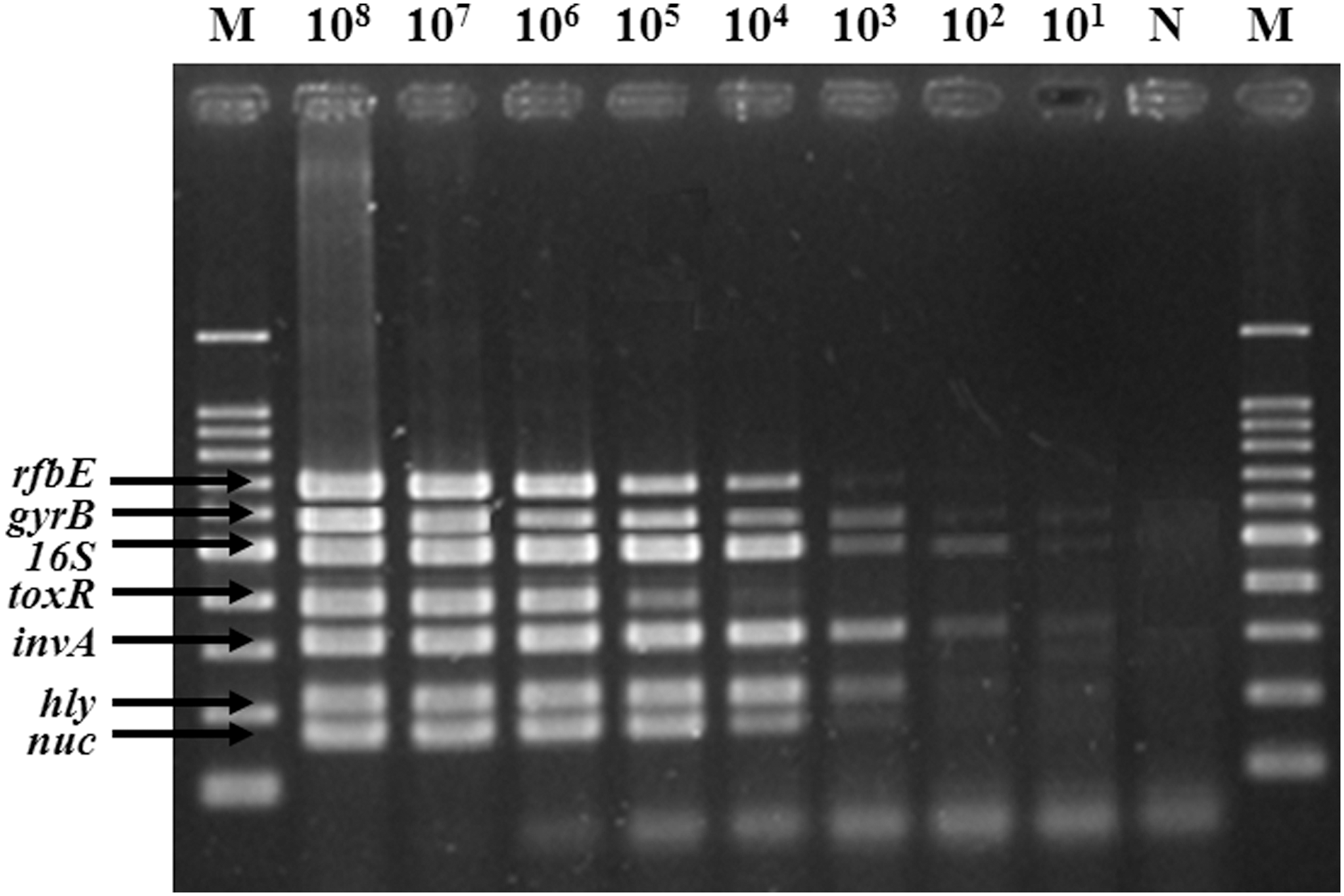
Sensitivity of the multiplex polymerase chain reaction assay on samples obtained from DNA extracts mix from serial dilutions of the six pathogens. M, 100-bp DNA ladder; N, negative control.
Evaluation of the multiplex PCR assay in artificially inoculated and natural food samples
In order to assess the detection sensitivity of the multiplex PCR assay for its application to food samples, fresh lettuce was inoculated with different concentrations of six foodborne pathogens (100–108 CFU/mL) and a noninoculated sample of lettuce was included as negative control. To improve the sensitivity and reproducibility of the multiplex PCR assay, the artificially inoculated lettuce and noninoculated lettuce were incubated for 4, 8, and 12 h in SEB enrichment medium. The DNA extracted from these samples was also tested using the multiplex PCR assay, in triplicate. The results (Fig. 3) demonstrated that after 8-h enrichment, the multiplex PCR assay was able to correctly identify the presence of the six foodborne pathogens at all different inoculated levels and down to the lowest concentration of 100 CFU/mL in lettuce. When 4-h instead of 8-h enrichment was used, all pathogens except L. monocytogenes were also detected.
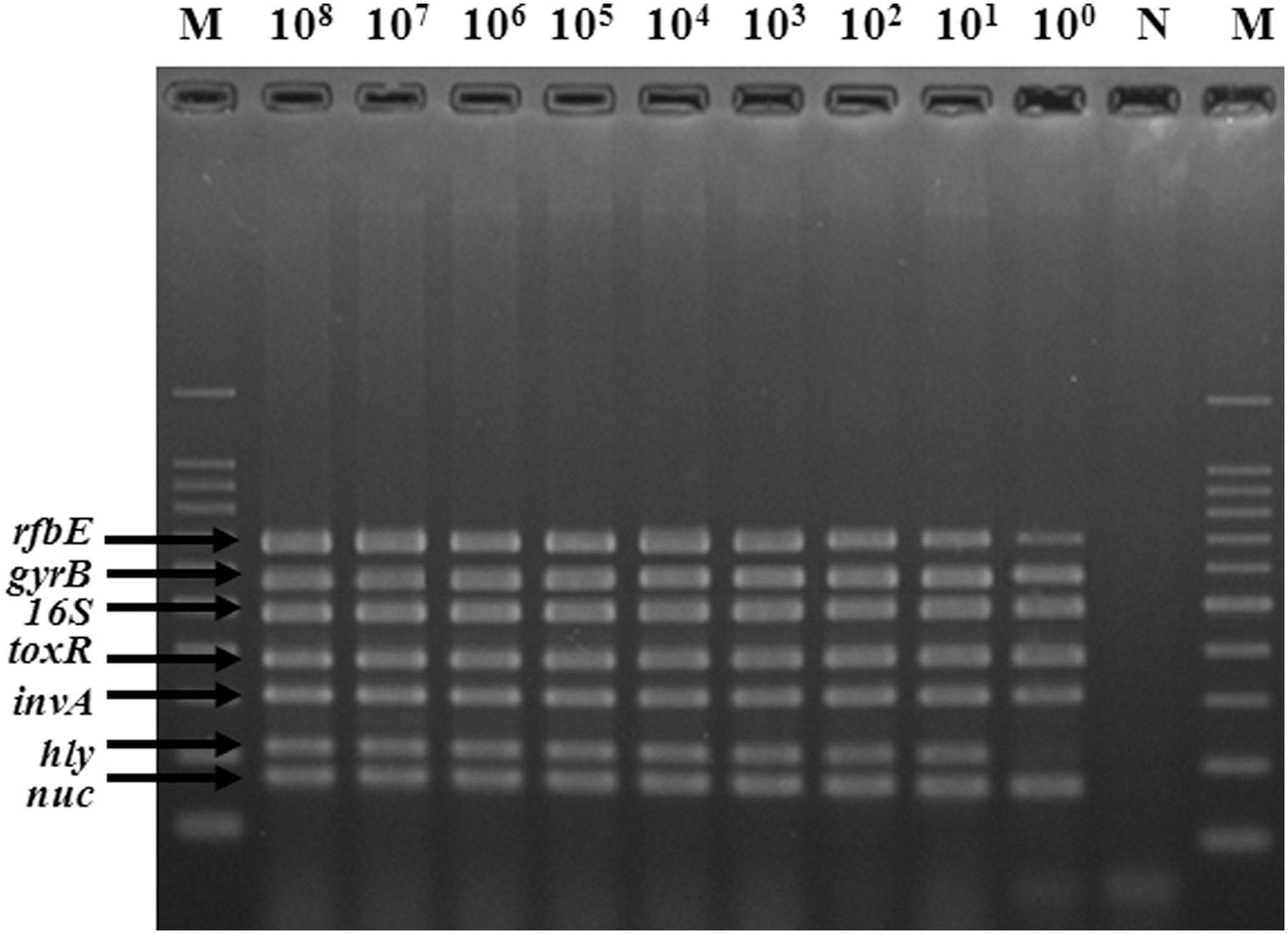
Application of the multiplex polymerase chain reaction assay to the detection of the target pathogens in fresh lettuces inoculated with different concentrations (100–108 colony-forming units mL−1) of the six pathogens mixture after 8-h enrichment. M, 100 bp DNA ladder; N, negative control.
To evaluate the validity of this multiplex PCR assay for simultaneous detection of pathogens in Korean ready-to-eat food, six foodborne pathogen mixtures were inoculated into samples of selected food (sandwiches, spicy chicken, steamed pork hocks, kimbab [rice rolled in dried seaweed with ingredients], sunsik [ground vegetables and grain powder], oyster, fresh tomato juice, sushi, and salad). Each sample was inoculated with an equal quantity of the six pathogen mixtures with different concentrations (100–102 CFU/mL). The artificially inoculated samples were cultured at 37°C for 8 h, and were analyzed using the multiplex PCR. However, the results were not consistent with the result in lettuce, where the lowest detection level such as 100 CFU/mL had been achieved after 8 h of enrichment against six foodborne pathogens. Although B. cereus, E. coli O157:H7, and S. aureus of six target pathogens could be detected from the selected food samples with inoculated levels of 100 CFU/mL, V. parahaemolyticus (toxR) and Salmonella spp. (invA) were not detected in the sandwich and spicy chicken samples with the 100 CFU/mL and in the kimbab and oyster samples with 100 and 101 CFU/mL. L. monocytogenes (hly) could not be detected in the oyster sample with 100 and 101 CFU/mL (Table 3). However, when a 12-h enrichment culture step was used, all bacteria were detected in all inoculated food samples.
In the previously reported multiplex PCR assays, Zhang et al. (2009) reported the highest sensitivity of 100 CFU/mL for four pathogens, such as E. coli O157:H7, L. monocytogenes, S. enterica, and S. aureus from eight artificially inoculated food samples after 24 h of enrichment. The multiplex PCR assay developed by Kim et al. (2007) also allowed for simultaneous detection at concentrations of 101 CFU/mL of five foodborne pathogens including E. coli O157:H7, L. monocytogenes, S. enterica, V. parahaemolyticus, and S. aureus in inoculated milk, pork, and chicken samples, after only 8 h of incubation time. The multiplex PCR assay established in this study was similar or more sensitive with the same incubation time when compared with Kim et al. (2007, 101 CFU/mL). It could also detect the six foodborne pathogens with the lowest level of 100 CFU/mL (Table 3) after 12 h of enrichment. Consequently, a 12-h enrichment period is required in this multiplex PCR assay for detecting food samples contaminated with a low level of foodborne pathogens.
To evaluate the practical use of our multiplex PCR assay, we compared this assay with the culture-based method for detecting six pathogens in 38 ready-to-eat foods. Of 23 kimbab samples tested, one sample was identified to contain both S. aureus and B. cereus by multiplex PCR. Among the 15 sandwich samples, one sample was found to be S. aureus–positive by multiplex PCR. E. coli O157:H7, L. monocytogenes, S. enterica, and V. parahaemolyticus could not be detected in any of these samples either by multiplex PCR or by culture-based detection. The same samples also yielded B. cereus and S. aureus following culture-based detection (not shown). Even though the naturally occurring food samples tested in the present study are low in number, the results obtained are valuable and highly promising and need to be evaluated further with diverse food samples.
The multiplex PCR assay has largely been applied to detect different species of several microbial niches in order to differentiate closely related species and to recognize single species. But cross-amplification reactions and false-positive signals may become a major concern when this technique is used as a defining method for differentiating foodborne pathogens in complex food matrices. Furthermore, inhibitors of the multiplex PCR can be found in microbial DNA solutions extracted from complex food such as kimbab. However, the multiplex PCR assay not only may replace the more labor-intensive conventional culture methods, but may also allow the detection of species that are present at low levels that can remain undetected by plating (Jamil et al., 1993; Settanni et al., 2007).
The multiplex PCR assay described here can simultaneously detect six foodborne pathogens of E. coli O157:H7, B. cereus, V. parahaemolyticus, Salmonella spp., L. monocytogenes, and S. aureus in Korean ready-to-eat food. It was found to be sufficient in specifically and simultaneously detecting as few as 100 CFU/mL of the six pathogens in artificially inoculated food samples after enrichment for 12 h. Also, it was successfully used in practical ready-to-eat food. Thus, the multiplex PCR assay developed in this study is an effective qualitative method to detect the six foodborne pathogens in many foods and will also be useful for the food industry and various regulatory agencies.
Footnotes
Acknowledgments
This work was partly supported by the Korea Food Research Institute and the IT R&D program of Ministry of Trade, Industry, and Energy (MOTIE)/Ministry of Science, ICT, and Future Planning (MSIP)/Korea Evaluation Institute of Industrial Technology (KEIT).
Disclosure Statement
No competing financial interests exist.