
Abstract
Listeria monocytogenes is an important foodborne pathogen commonly isolated from food processing environments and food products. This organism can multiply at refrigeration temperatures, form biofilms on different materials and under various conditions, resist a range of environmental stresses, and contaminate food products by cross-contamination. L. monocytogenes is recognized as the causative agent of listeriosis, a serious disease that affects mainly individuals from high-risk groups, such as pregnant women, newborns, the elderly, and immunocompromised individuals. Listeriosis can be considered a disease that has emerged along with changing eating habits and large-scale industrial food processing. This disease causes losses of billions of dollars every year with recalls of contaminated foods and patient medical treatment expenses. In addition to the immune status of the host and the infecting dose, the virulence potential of each strain is crucial for the development of disease symptoms. While many isolates are naturally virulent, other isolates are avirulent and unable to cause disease; this may vary according to the presence of molecular determinants associated with virulence. In the last decade, the characterization of genetic profiles through the use of molecular methods has helped track and demonstrate the genetic diversity among L. monocytogenes isolates obtained from various sources. The purposes of this review were to summarize the main methods used for isolation, identification, and typing of L. monocytogenes and also describe its most relevant virulence characteristics.
Introduction
L
Some groups have a higher risk to develop the disease, such as newborns, pregnant women, the elderly, transplant patients, those with immunodeficiency, including patients with human immunodeficiency virus, and those diagnosed with cancer. Although listeriosis is relatively rare, it is considered a serious disease. L. monocytogenes is responsible for 19% of total deaths associated with the consumption of contaminated food in the United States (Scallan et al., 2011).
Many factors make this foodborne pathogen a public health concern. These include changes in industrial processes highlighted as relevant for L. monocytogenes, such as the use of refrigeration systems, large-scale industrial food processing, and changes in eating habits of the population toward consumption of ready-to-eat (RTE) products (Jay et al., 2005; Swaminathan and Gerner-Smidt, 2007). Several studies have been published, mainly in industrialized countries, to understand the mechanisms of L. monocytogenes persistence in the food processing environment, its contamination routes, and its potential to cause disease in the host.
In the last few decades, phenotypic and genotypic studies have led to new insights into the genetic evolution and virulence potential of Listeria spp. This characterization has been possible due to the development of accurate techniques for typing of this pathogen. In this study, we present an updated description of the main methods described for isolation and identification of L. monocytogenes, the use of typing methods for surveillance and outbreak tracking, and also the most pathogenic features and its importance for each step of the virulence cycle.
Isolation Methods and Species Identification
To date, 17 species and 6 subspecies have been described for the genus Listeria (
Conventional microbiological methods are indispensable for the isolation and identification of Listeria spp. from environmental sources, as well as from food products and clinical samples. Standard methods for isolation from different sources have been described by the International Organization for Standardization (ISO), the U.S. Food and Drug Administration (FDA), International Dairy Federation (IDF), U.S. Department of Agriculture—Food Safety and Inspection Service (USDA-FSIS), and Health Protection Branch (HPB) (Ryser and Marth, 2007).
For all methods, samples are preincubated in basal medium for the recovery of injured cells, followed by enrichment in selective/differential supplemented broths, such as Buffered Listeria Enrichment Broth, Half-Fraser Broth (HFB), and Fraser Broth. After the primary selective enrichment, the obtained cultures are streaked on selective agars; the most common culture media used in this step are the PALCAM Agar Base (Listeria spp. develop small brown/black colonies surrounded by black halos due to esculin hydrolysis), Listeria Selective Agar (Oxford, with the same characteristic colonies observed as in PALCAM), and the selective Chromogenic Listeria Agar (ALOA, in which Listeria spp. develop blue/green colonies, and L. monocytogenes presents an additional feature: an opaque white halo due to its lecithinase activity).
Alternative enrichment broths and culture media can also be considered, and indications may vary in accordance with the adopted protocol (Table 1) (Farber et al., 1994; AOAC, 1996, 2000; Scotter et al., 2001a, b; Hitchins and Jinneman, 2011; USDA, 2013). After isolation, when incubated in trypticase soy agar, pure cultures appear as nonpigmented and translucent and present bluish color when viewed under oblique lighting (Jay et al., 2005).
ALOA, Agar Listeria Ottaviani and Agosti Medium; AOAC, Official Methods of Analysis; BAM, Bacteriological Analytical Manual; BCM, Biosynth Chromogenic Medium; BLEB, Buffered Listeria Enrichment Broth; ES, environmental samples; FB, Fraser Broth; FDA, Food and Drug Administration; HFB, Half-Fraser Broth; HPB, Health Protection Branch; IDF, International Dairy Federation; ISO, International Organization for Standardization; LiCl, lithium chloride; LPM, Lithium Chloride–Phenylethanol–Moxalactam Medium; mFB, modified Fraser Broth; MOPS-BLEB, Morpholine-Propanesulfonic Acid–Buffered Listeria Enrichment Broth; MOX, Modified Oxford Listeria Selective Agar; ONE, Oxoid Novel Enrichment Broth; OXA, Oxford Medium; PALCAM, PALCAM Listeria Selective Agar; USDA-FSIS, U.S. Department of Agriculture—Food Safety and Inspection Service; UVM, Modified University of Vermont Broth.
The biochemical species identification is possible by the catalase reaction, carbohydrate fermentation profile, hemolytic activity, and other complementary tests, such as Gram staining and motility test at 25°C. Commercial kits, as the API Listeria test kit (bioMérieux, Marcy-l′Etoile, France), have been designed for the genus Listeria, allowing easy biochemical differentiation in a microtube format, which targets the presence or absence of arylamidase (DIM test), hydrolysis of esculin, presence of α-mannosidase, and acid production from
To improve the assessment of hemolysis, and ensuring a more reliable differentiation between the hemolytic species L. monocytogenes, L. ivanovii, and Listeria seeligeri, various authors recommend the use of the CAMP (Christie, Atkins, and Munch-Petersen) test (Seeliger and Jones, 1986). The CAMP test detects synergistic hemolysin activity of Listeria spp., with the beta-toxin of Staphylococcus aureus and the exofactor of Rhodococcus equi, on a sheep blood agar plate. L. monocytogenes has a positive reaction with S. aureus but a negative one with R. equi, and L. ivanovii displays the reverse results. Even more so, L. seeligeri can show a weak positive reaction with S. aureus. Therefore, skill is necessary to perform and interpret these results (Allerberger, 2003).
L. monocytogenes can be identified by immune-based techniques, such as enzyme-linked immunosorbent assays (ELISAs) and immunomagnetic separation, but these methods have a minimum detection limit of 105 cells/mL for bacterial detection from environmental samples by antigen–antibody reaction.
It is important to point out that in recent years, several new species of Listeria have been described, and many of them have their own characteristics, distinct from other species. Considering this, molecular-based methods, using specific primers, and genetic sequencing are required to properly perform phylogenetic position analysis and species attribution (Liu, 2006; Weller et al., 2015). The polymerase chain reaction (PCR) has been widely used for Listeria identification. Many primers were designed for differentiation of Listeria spp., and the most common targets to detect L. monocytogenes include the virulence-associated genes hly, actA, plcA, plcB, inlA, inlB, inlC, and inlJ (Liu, 2006). The PCR procedure has proved to be a rapid and sensitive method for the routine analysis of different types of food (Aznar and Alarcón, 2003).
According to Kaclıková et al. (2003), L. monocytogenes can be identified by PCR equivalent to ISO 11290-1 or ISO 10560 in terms of detection limit (100 colony-forming units [CFUs]/25 or 10 g) and with 100% relative accuracy in cheese, smoked fish, and RTE meat products. Currently, several multiplex PCR protocols are being used for the rapid detection of Listeria spp. and potentially pathogenic L. monocytogenes from different sources. Rawool et al. (2007) developed a multiplex PCR to detect the four virulence-associated genes plcA, hlyA, actA, and iap in enrichment milk samples using previously described primers. Liu et al. (2007) standardized a multiplex PCR to detect the inlA, inlC, and inlJ genes that also provide identification regarding virulence potential.
Ryu et al. (2013) developed a multiplex PCR for the rapid and simultaneous detection of six Listeria spp. (L. grayi, L. innocua, L. ivanovii, L. monocytogenes, L. seeligeri, and L. welshimeri) from meat processed foods. The authors suggested that this method can be useful for the detection of Listeria spp. in contaminated foods and clinical samples. Recently, Liu et al. (2015) developed a new multiplex PCR method, which is able to discriminate among L. monocytogenes, L. innocua, L. seeligeri, L. welshimeri, L. ivanovii, and L. grayi in deli meats. This method allowed accurate detection of Listeria spp., but pre-enrichment procedures are important to increase sensitivity.
Furthermore, real-time PCR-based methods have been developed for the rapid identification and quantification of Listeria spp. from a variety of sources (Le Monnier et al., 2011; Barbau-Piednoir et al., 2013; Gianfranceschi et al., 2014; Quero et al., 2014). Real-time PCR was designed for the diagnosis of meningoencephalitis caused by L. monocytogenes using the hly gene target, and it provided reproducible results over a wide range of concentrations (Le Monnier et al., 2011).
Recently in Europe, a study was conducted at 12 laboratories in 6 countries using real-time PCR-based methods for L. monocytogenes detection in soft cheese, and the results were compared with those of the ISO 11290-1 standard method. The observed limit of detection after pre-enrichment in HFB by real-time PCR was down to 10 CFUs per 25 g of sample. The excellent agreement observed among the laboratories suggests an option soon to be implemented by the authorities and the food industry (Gianfranceschi et al., 2014). However, the equipment necessary for this analysis is still not available in many laboratories. Mass spectrometry–based methods for identification and typing of Listeria spp. were already described and allow differentiation of pathogenic strains and even clonal lineages (Barbuddhe et al., 2008).
In addition, sequencing of the 16S rRNA gene, 23S rRNA gene, or iap gene has been used by many authors for accurate phylogenetic analysis and species identification (Liu, 2006). Genetic sequencing has become more popular in the last decade, and currently, there are several public and private companies providing these services at relatively low cost.
L. monocytogenes typing
Typing methods are useful for tracking the origins of the pathogen and to characterize its virulence potential. The typing can be performed by phenotypic and molecular methods, and several methods have been described in last decades for epidemiological investigations and genetic characterization (Boerlin and Piffaretti, 1991; Brosch et al., 1994; Schönberg et al., 1996; Wernars et al., 1996; Salcedo et al., 2003; Doumith et al., 2004). In addition to the epidemiological data, the combination of different typing methods provides a proper characterization of isolates obtained from different sources.
L. monocytogenes strains are grouped into four phylogenetic lineages, which can be differentiated by conventional and molecular techniques. The serotypes 1/2b, 3b, 3c, and 4b constitute the lineage I; serotypes 1/2a, 1/2c, and 3a constitute the lineage II, and rarely, serotypes 4a, 4c, and atypical 4b form the lineage III. Recently, a new lineage was described (lineage IV), which is also represented by serotypes 4a, 4c, and atypical 4b but rarely found (Table 2) (Ward et al., 2008; Orsi et al., 2011).
One of the first phenotypic techniques used to type L. monocytogenes was serotyping (Seeliger and Höhne, 1979). Based on the expression of surface proteins, namely somatic antigens (O factor) and flagellar antigens (H factor), 13 L. monocytogenes serotypes have been described (1/2a, 1/2b, 1/2c, 3a, 3b, 3c, 4a, 4ab, 4b, 4c, 4d, 4e, and 7) (Allerberger, 2003; Seeliger and Höhne, 1979). Serotypes 1/2a, 1/2b, and 4b are involved in 95% of all listeriosis cases, with serotype 4b responsible for higher hospitalization rates and deaths (Swaminathan and Gerner-Smidt, 2007; Cartwright et al., 2013).
The conventional agglutination method (Seeliger and Höhne, 1979; Seeliger and Jones, 1986) is the reference protocol for serotyping L. monocytogenes isolates obtained from clinical and food samples; however, it is time consuming and expensive. Palumbo et al. (2003) developed an ELISA-based assay serotyping L. monocytogenes; however, it was also time consuming, and it was still dependent on producing high-quality antisera.
Due to practical reasons, molecular methodologies have been widely used for the rapid screening of L. monocytogenes strains based on serogroup divisions. These methods provide rapid and low-cost results, but the exact identification of the serotype is not possible since these protocols propose a serogroup categorization that includes different serotypes, usually the most prevalent serotype and other nonfrequent serotypes (Borucki and Call, 2003; Doumith et al., 2004; Zhang and Knabel, 2005; Kérouanton et al., 2010; Vitullo et al., 2013).
Serogrouping proposed by Doumith et al. (2004) differentiates L. monocytogenes into four molecular serogroups: IIa corresponded to serotypes 1/2a and 3a; IIc to 1/2c and 3c; IIb to 1/2b, 3b, and 7; and IVb to 4b, 4d, and 4e (Doumith et al., 2005). This is now the main protocol considered for serogrouping isolates obtained from food and clinical samples. However, various similar PCR-based methods have been described in the last decade (Zhang and Knabel, 2005; Chen and Knabel, 2007; Kérouanton et al., 2010; Vitullo et al., 2013), and all of them propose a similar serogroup categorization.
Recently, Nho et al. (2015) developed a new multiplex PCR to differentiate serotypes 1/2a, 1/2c, 3a, and 3c, but few isolates have been tested. The great advantage of these methods is to obtain fast results at low cost; however, some isolates from serotypes 1/2a and 4b may present atypical profiles, as described before (Kérouanton et al., 2010; Leclercq et al., 2011; den Bakker et al., 2014). This problem is related to the target genes in the protocol, which are not serotype-specific surface antigens (Kérouanton et al., 2010; Huang et al., 2011; Leclercq et al., 2011; Lee et al., 2012; Camargo et al., 2016).
Phage typing is an alternative method for clustering L. monocytogenes, and it has also helped link listeriosis as a foodborne disease (Fleming et al., 1985). A number of phage sets have been developed, and typable strains vary from 52% to 78%, according to the number of phages used in these investigations and the susceptibility of each strain (Audurier and Martin, 1989; Van Der Mee-Marquet et al., 1997).
A significant number of Listeria strains are untypable by this technique, limiting its scope in L. monocytogenes typing. In contrast, in recent years, its application has been further explored, how being considered as a biocontrol tool with foods (Chibeu et al., 2013; Oliveira et al., 2014), diagnostics (Hagens et al., 2011; Tolba et al., 2012), in immobilization and detection (Habann et al., 2014), and as an antimicrobial (Schmelcher et al., 2012). According to Hagens and Loessner (2014), phages that are specific for Listeria provide numerous novel tools for various interesting approaches.
Although phenotypic methods have been useful for typing L. monocytogenes, currently, molecular methods appear to be more reliable and sensitive, and these tools are widely used worldwide. Molecular typing methods can be based on the use of restriction enzymes. Ribotyping method is based on the restriction fragment length polymorphisms (RFLPs) associated with the ribosomal operons, and it was used by Wiedmann et al. (1997) for lineage-specific differentiation of L. monocytogenes isolates and to also assess their pathogenic potential. Ribotyping has been successfully used to type L. monocytogenes from various sources, offering the best discriminatory power when associated with serotyping (Kabuki et al., 2004; De Cesare et al., 2007; Sant'Ana et al., 2012).
According to Wiedmann (2002), the use of enzymes EcoRI and PvuII by two distinct restriction reactions provides better strain discrimination. However, it cannot efficiently differentiate strains from serotypes 1/2b and 4b, thus limiting its use for epidemiological investigations (Jadhav et al., 2012). Its discriminatory power is similar to multilocus enzyme electrophoresis (MLEE). MLEE divides the L. monocytogenes into two primary subgroups: one represented by serotypes 1/2b, 4a, and 4b and another by serotypes 1/2a and 1/2c. The bacterial isolates are differentiated according to the electrophoretic mobility variation of a large number of metabolic enzymes. However, this method is labor intensive, suffers from interlaboratory nonreproducibility, and presents less discriminatory power compared with other molecular methods (Boerlin and Piffaretti, 1991; Ryser and Marth, 2007).
Pulsed-field gel electrophoresis (PFGE) is widely used for genetic characterization of microorganisms, and the method standardized for L. monocytogenes uses a combination of two restriction enzymes: ApaI and AscI (Graves and Swaminathan, 2001). This method is based on RFLPs of bacterial DNA, resulting for L. monocytogenes fragments ranging from 30 to 600 kb, which appears in 6–25 bands in agarose gel electrophoresis (Graves and Swaminathan, 2001; Liu, 2006; Jadhav et al., 2012). PFGE is widely used and considered the gold standard to track L. monocytogenes from food processing facilities, foods, and clinical samples. The CDC and Association of Public Health Laboratories have created a network for tracking L. monocytogenes in epidemiological studies and foodborne outbreaks using PFGE as a standardized method for subtyping this pathogen.
The European Center for Disease Prevention and Control (ECDC) also uses the PFGE as a standard procedure for typing L. monocytogenes isolates. PFGE is more discriminatory than serotyping and other methods based on restriction enzymes, and it shows the possibility that cultures belonging to the same serotype have different genetic profiles (Graves and Swaminathan, 2001; Gudmundsdóttir et al., 2005; Fugett et al., 2007; Galvão et al., 2012; Hächler et al., 2013). The analysis of genetic band profiles is done by a computer software, which enables a quick and easy comparison of the data and may involve the profiles of isolates obtained from food, with profiles of strains involved in outbreaks. This method is useful for epidemiological studies, implementation of corrective actions, and monitoring of critical control points. Major disadvantages of PFGE are the time required to complete the procedure (minimum 24 h), high costs of restriction endonucleases, and specialized equipment for electrophoresis.
Several PCR-based methods also have been used as an alternative for typing L. monocytogenes strains, such as molecular serogrouping (previously described), random amplified polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP), PCR-RFLP, repetitive element PCR (REP-PCR), fluorescence amplified fragment length polymorphism (fAFLP), and multilocus variable-number of tandem-repeat analysis (MLVA). In addition, DNA sequencing–based subtyping techniques, as multilocus sequence typing (MLST), multivirulence-locus sequence typing (MVLST), and single-nucleotide polymorphism (SNP) analysis, have been demonstrated (Liu, 2006; Jadhav et al., 2012).
The RAPD method allows a single arbitrarily selected primer, generally 10 bp in length, to anneal with complementary sequences on the target DNA, creating a genetic profile (Lawrence et al., 1993). This protocol has been used for fast typing of L. monocytogenes from foods and can provide valuable information about possible persistence of strains, also indicating potential sources of cross-contamination when used combined with serotyping (Vogel et al., 2001; Aurora et al., 2009). In addition, according to Hadjilouka et al. (2014), the combination of REP-PCR with any primer used in the RAPD analysis can provide an optimum discrimination of strains, but alone, the technique does not have high discriminatory power.
In the AFLP method, the total DNA is subject to digestion by two restriction enzymes, fragments are amplified using PCR, and the products are subjected to gel electrophoresis. The described protocols were based on different restriction enzymes, being BamHI and EcoRI for AFLP I and HindIII and HhaI for AFLP II. According to Parisi et al. (2010), AFLP and MLST methods produced similar results in terms of discriminating power in L. monocytogenes from food and environment, generating 62 and 66 types from isolates used in the study, respectively. The authors suggest that these two methods can be associated to provide high-resolution results. Lomonaco et al. (2011) compared AFLP and PFGE methods. Both protocols grouped L. monocytogenes strains into two main clusters, and the results showed a similar discriminatory power. According to the authors, AFLP can be successfully used to type L. monocytogenes strains obtained from foods and food processing facilities where PFGE is not available; however, it is also expensive and time consuming.
PCR-RFLP consists of PCR amplification of L. monocytogenes housekeeping or virulence genes, followed by digestion with selected restriction enzymes (HhaI, SacI, or HinfI) and separation by gel electrophoresis. The distinct band patterns allow differentiation among L. monocytogenes subtypes (Wiedmann et al., 1997). As advantages, this method requires only a small amount of starting DNA and may be used in epidemiological investigations to track L. monocytogenes (Liu, 2006). Strydom et al. (2013) subjected L. monocytogenes isolates to PCR-RFLP and PFGE, and the results indicated that both molecular subtyping methods were sensitive and specific enough to assess the genetic diversity.
REP-PCR targets dispersed repetitive sequence elements, such as repetitive extragenic palindromes of 35–40 bp, which are found in the extragenic regions of the genome in direct or reverse orientation. The repetitive extragenic palindromes sequences represent useful single primer set binding sites for PCR amplification, and different locations of these sequences in the genome produce variable fragment sizes. According to Harvey et al. (2004), PFGE presented greater discriminatory power for typing L. monocytogenes isolates from foods than REP-PCR and multilocus enzyme electrophoresis, although the three methods were able to differentiate closely related L. monocytogenes strains. Hadjilouka et al. (2014) subjected 121 strains from food products to RAPD and REP-PCR, and the last method provided better differentiation among isolates. Apparently, REP-PCR possesses a discriminatory power similar to PFGE and ribotyping; as REP-PCR is faster and cheaper than these typing techniques, it can be considered as an important alternative for typing L. monocytogenes.
fAFLP is a variant of AFLP, done using fluorescent PCR primers. fAFLP is done by a modified protocol previously described for typing Campylobacter. By this protocol, Listeria genomic DNA is digested using the enzyme pair HindIII and HhaI. Roussel et al. (2013) showed that 109 L. monocytogenes isolates from human clinical cases, foods, and food processing environments as well as animal cases, reference strains, and isolates associated with outbreaks and sporadic cases were divided by fAFLP and PFGE into three clearly distinguishable lineages, and both methods showed equal discriminatory power. The authors suggested that fAFLP is a good alternative to PFGE for L. monocytogenes typing, and it can be used for investigations of listeriosis outbreaks and tracking contamination sources in food processing environments.
MLVA focuses on the study of the variability of the number of tandem repeats (VNTRs) at specific loci of bacterial genomes. VNTRs are short segments of DNA that have variable copy numbers, and the difference in copy numbers at specific loci is used to measure relationships among strains. The primers are designed from the flanking regions of VNTRs. The PCR products may be separated on agarose gels (according to sizing of individual fragments, the copy numbers are detected) and/or subjected to DNA sequencing systems (Murphy et al., 2007; Sperry et al., 2008).
Chenal-Francisque et al. (2013) evaluated 18 VNTRs loci and combined the 11 best ones into 2 multiplex PCR assays. The authors suggest that this protocol is useful for the characterization of L. monocytogenes strains and that it represents an attractive first-line screening method to epidemiological investigations and listeriosis surveillance. Furthermore, it is of low cost and easy to perform and provides rapid results (around 8 h), with portable (numerical) results. However, this method exhibits lower discriminatory power than PFGE based on ApaI and AscI restriction enzymes, as observed by Sperry et al. (2008).
According to Lindstedt et al. (2008), MLVA was slightly more discriminatory than PFGE for isolates from Norway, consisting of 28 MLVA profiles and 24 PFGE profiles. The opposite was observed for isolates from Sweden, producing 42 MLVA profiles and 43 PFGE profiles; however, only AscI enzyme was used for PFGE in this study. In addition, Chen et al. (2011) reported the MLVA application for typing L. monocytogenes directly in food samples. This method successfully typed strains from cheese, roast beef, egg salad, and vegetable samples after a 48-h enrichment, generating accurate, reproducible, and high-quality results from artificially contaminated food samples.
Many of the techniques described above have been widely used in phenotypic and genotypic studies and led to new insights into ecology, epidemiology, virulence potential, and genetic evolution. In the last decade, PCR-based sequencing methods have become available for many laboratories, providing several advantages, including electronic portability of nucleotide sequences, which allows for quick comparisons between laboratories.
One of the first PCR-based sequencing techniques described for typing pathogenic microorganisms was MLST (Maiden et al., 1998). Using this technique, it is possible to evaluate the DNA sequence variations of housekeeping genes and compare the sequences with their original profiles. For each gene, different sequences found are assigned as alleles, and these provide a profile that defines unambiguously the type of sequence of each isolate. Changes in the nucleotide housekeeping genes occur relatively slowly, making this method suitable for epidemiological investigations (Enright and Spratt, 1999; Cooper and Feil, 2004).
L. monocytogenes typing by MLST was first demonstrated by Salcedo et al. (2003). Nowadays, a protocol adapted by Ragon et al. (2008) has become widely used to characterize L. monocytogenes isolates, targeting seven housekeeping genes: acbZ (ABC transporter), bglA (beta-glucosidase), cat (catalase), dapE (succinyl diaminopimelate desuccinylase), dat (
In addition, Zhang et al. (2004) developed an MVLST method that exhibits higher discriminatory power than ribotyping, PFGE, and MLST. This method is based on the sequencing of three virulence genes (prfA, inlB, and inlC) and three virulence-associated genes (dal, lisR, and clpP), and it can improve the discriminatory power of MLST while also providing information about virulence potential of L. monocytogenes strains. In addition, the inclusion of the two virulence genes (inlA and actA) was proposed by Chen et al. (2007) to complement the six previous genes used in the analysis. Recently, various studies have used MLST and MVLST combined and demonstrated success for epidemiological investigations and also genetic/virulence evolution (Knabel et al., 2012; Cantinelli et al., 2013; Martín et al., 2014; Yin et al., 2015).
Another approach is to type L. monocytogenes by assessing SNPs at multiple locations on the genome. Unnerstad et al. (2001) categorized 106 L. monocytogenes on the basis of SNPs in the inlB gene, and strains were clustered into four groups (serogroups 1/2a and 1/2c were clustered in one group, 1/2b and 3b in another group, and serotype 4b strains in two groups). Ducey et al. (2007) designed multilocus genotyping (MLGT) by sequencing 23,251 bp of DNA from 22 genes distributed across 7 genomic regions to efficiently target SNP variation and type L. monocytogenes isolates from lineage I. This protocol also provides information about a specific virulence-attenuated subtype with a characteristic truncation mutation in inlA. Considering this, MLGT represents a significant new tool for pathogen surveillance, risk assessment, outbreak detection, and epidemiological investigations (Ducey et al., 2007; Ward et al., 2008).
Since the genomes of L. monocytogenes and L. innocua were published by Glaser et al. (2001), various studies have been focused on comparative genomics to understand the genetic relationships among L. monocytogenes lineages and molecular mechanisms associated with virulence potential (Nelson et al., 2004; Gilmour et al., 2010; Bécavin et al., 2014).
New technologies, as next-generation sequencing (post-Sanger sequencing technologies) using Illumina MiSeq or NextSeq platforms, have been described and successfully applied for extremely fast whole-genome sequencing. To accurate data analysis, new bioinformatic devices are facilitating comparative genomics and data sharing among researchers, which improves the quality of epidemiological studies (Stasiewicz et al., 2015; Bergholz et al., 2016). These studies indicate that in a very near future, genome sequencing will replace many of the previously described methods due to its discriminatory power and portability of generated data (Kwong et al., 2016).
Pathogenicity Factors and Virulence Potential
L. monocytogenes has acquired a variety of virulence factors that allow it to successfully invade and survive within host cells. This bacterium has a characteristic way to colonize the human host using phagocytic cells to be distributed in the body for cell-to-cell spread (Fig. 1) (Chaturongakul et al., 2008). After adhesion and invasion of the intestinal epithelium, L. monocytogenes spreads throughout the body, infecting macrophages, epithelial cells, endothelial cells, hepatocytes, fibroblasts, and cells of the nervous system (Fig. 2). Translocation from the lumen into the intestinal epithelial cells is mediated by the surface-associated virulence protein internalin A (InlA) encoded by the gene inlA; these proteins interact with specific receptors on host cells, leading to receptor-mediated internalization. InlA protein binds to glycoprotein E-cadherin present on the host cell, resulting in a rearrangement of the cell cytoskeleton and entry of the pathogen into the host cell (de las Heras et al., 2011).
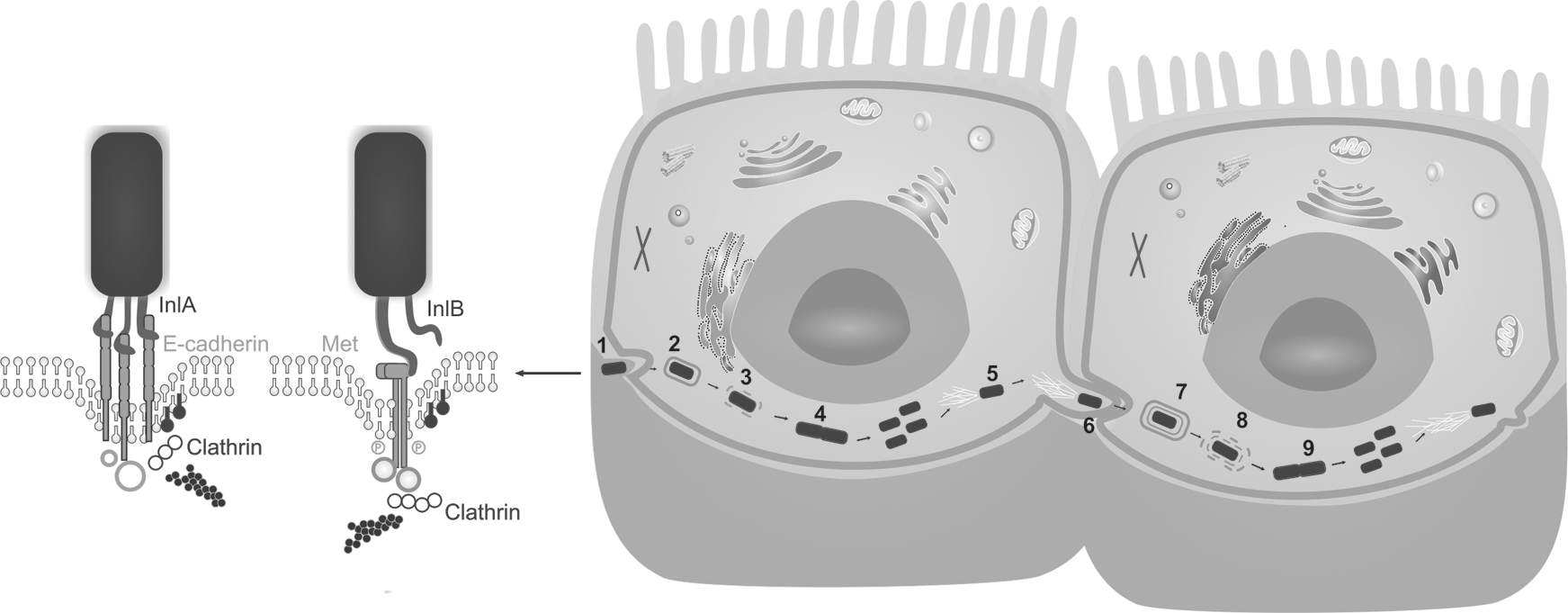
Steps of Listeria monocytogenes intracellular infection cycle: (1) attachment to the host cell surface and internalization via a “zipper mechanism” is promoted by two listerial surface proteins (InlA and InlB, encoded by inlA and inlB genes) with their respective cell surface receptors (E-cadherin and the receptor tyrosine kinase c-Met); (2) this results in engulfment of bacteria and endosome formation; (3) the escape from the vacuole is mediated by three membrane-damaging factors: a pore-forming toxin LLO, encoded by the gene hly, and two phospholipases PlcA and PlcB, encoded by genes plcA and plcB (the latter needs to be processed by the Mpl metalloprotease after secretion, which is encoded by mpl gene); (4) replication in the cell cytosol using cytosolic resources (Listeria adapt their metabolism and start the expression of several genes, such as the hexose transporter Hpt or the lipoate protein ligase LplA1, for multiplication); (5) cell-to-cell spread mechanism, which allows intracellular motility, is mediated by the surface protein ActA (encoded by the gene actA), which induces actin polymerization, providing Listeria motility through the cytoplasm; (6) at the end of this cycle, Listeria reach the plasma membrane and induce the formation of pseudopod-like structures (listeriopods), allowing the invasion of the neighbor cell: this activity is mediated by InlC protein (encoded by inlC gene); (7) phagocytosis and formation of a double-membrane vacuole; and (8) rupture of the two-membrane vacuole and Listeria escape initiate a (9) new round of proliferation, actin-based motility, and intercellular spread. LLO, listeriolysin O.
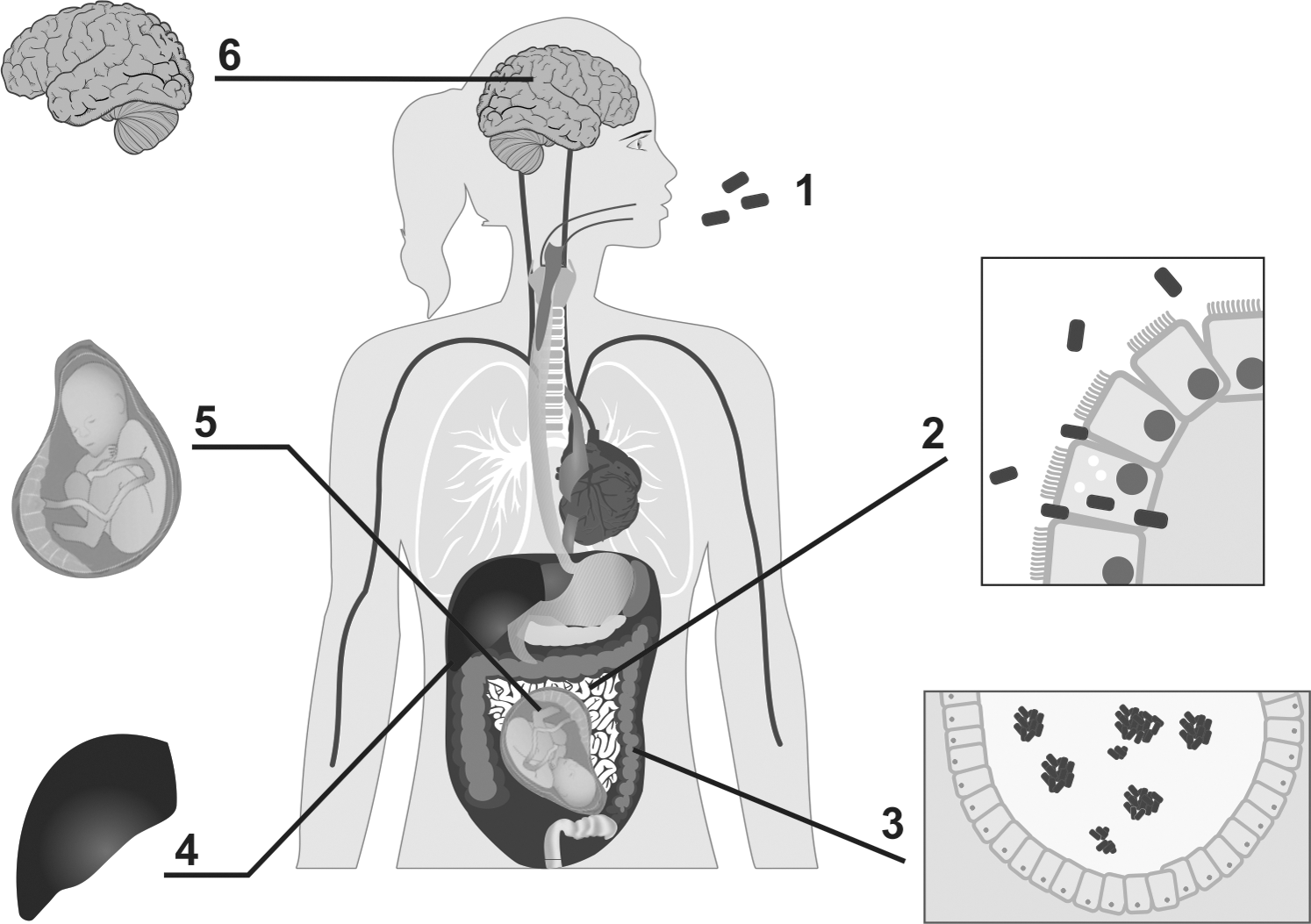
Human infection by Listeria monocytogenes: (1) ingestion of contaminated food; (2) Listeria colonization of the digestive tract can lead to symptoms, including diarrhea and fever; the bacteria cross the intestinal barrier, due to the production of InlA, reaching the mesenteric lymph nodes and accessing the circulatory system; (3) Listeria can form aggregates, ActA dependent, favoring the long-term persistence in the cecum lumen; (4) the first organs affected are the liver and spleen (which are reservoirs of bacterial persistence): these organs release this bacterium into the blood system, resulting in septicemia; (5) L. monocytogenes may cross the placental barrier leading to abortion or generalized neonatal infection (causing pneumonia, sepsis, or meningitis); and (6) in addition, L. monocytogenes can cross the blood–brain barrier and reach the brain, causing meningitis, meningoencephalitis, and rhombencephalitis.
After internalization, other virulence proteins, such as pore-forming cytolysin listeriolysin O (LLO) encoded by the gene hly and 2 phospholipases C (PlcA and PlcB) encoded by the genes plcA and plcB, help the bacterium to escape from phagosomes formed during the invasion process. The protein encoded by the hly gene causes pores in the membrane of the phagosome, resulting in lysis of the membrane and bacterial escape.
Furthermore, this protein is responsible for hemolytic activity in virulent isolates. The proteins encoded by the genes plcA and plcB operate in synergy within the vacuole, being activated by a metalloprotease (Mpl), which is encoded by mpl gene. For replication in the host cell cytoplasm, sugar phosphate permease HPT protein is required. Then, the spreading to neighboring cells is fully dependent on the actin polymerizing protein ActA, encoded by the gene actA, that guarantees the movement through the cell by induction of actin polymerization (Fig. 1). In addition, the actA protein also participates in the maintenance of Listeria aggregates in the cecum lumen (Vázquez-Boland et al., 2001; de las Heras et al., 2011).
L. monocytogenes can spread by the bloodstream to mesenteric lymph nodes, liver, spleen and multiply within host cells as well. Several genes encoding proteins related virulence, such as actA, plcA, plcB, hlyA, mpl, and prfA, that are located in the Listeria pathogenicity island 1 (LIPI-1) (Schmidt and Hensel, 2004), which is regulated by the protein PrfA. To avoid unnecessary expression in the environment, the PrfA regulon is selectively activated during infection (de las Heras et al., 2011).
Another important virulence-associated locus comprises the inlAB gene locus. The L. monocytogenes genome encodes 27 proteins now known as a family of internalins; however, InlA and InlB are the only internalins that have been directly implicated in host cell internalizations (Pizarro-Cerdá et al., 2012). In addition, the Listeria genomic island 1 (LGI1), initially identified by Gilmour et al. (2010), consists of genes that encode putative translocation, resistance, and regulatory determinants. These characteristics suggest that this locus plays an important role in bacterial persistence and/or pathogenicity. The internalin protein encoded by the gene inlA is critical to the invasion of the bacteria into the intestinal barrier during the initial stage of infection (Bonazzi and Cossart, 2006).
Several studies have shown that mutations in the inlA gene can lead to premature stop codons (PMSCs), resulting in the expression of a nonfunctional truncated protein (Tamburro et al., 2010). Although PMSCs are found in isolates from lineages I and II, they are most commonly observed in isolates from lineage II (serotypes 1/2a and 1/2c) that are frequently associated with environmental and food sources.
In a study conducted by Van Stelten et al. (2010), it was confirmed that mutations in isolates from foods are more common than those from clinical cases. Associated with this, strains with mutations had reduced invasion of Caco-2 cells, consistent with the critical role of internalins in the infection process. In this way, sequencing data of the inlA gene provide important information regarding virulence potential of L. monocytogenes strains obtained from the food production chain and clinical cases. In addition, sequencing the inlA gene helps estimate the risk of exposure to pathogenic strains through the consumption of food (Olier et al., 2005; Rousseaux et al., 2004; Van Stelten et al., 2010).
Several factors may contribute to the development of listeriosis, such as the number of ingested bacterial cells, host immunity, and virulence potential of each strain. Despite being naturally virulent, some L. monocytogenes strains are avirulent and unable to cause disease. The evaluation of the virulence potential should be made by appropriate laboratory methods, such as sequencing of virulence genes and evaluation of the expression of virulence factors and their effects on host cell invasion through cell culture studies (Liu, 2006). Such assessment may contribute to a better understanding of the virulence potential of strains isolated from the food production chain and clinical cases, and it provides input to implement programs to promote the prevention of listeriosis.
Concluding Remarks
The intracellular foodborne pathogen L. monocytogenes is ubiquitous in nature. Genetic profiles obtained by molecular methods have helped demonstrate the genetic diversity of this foodborne pathogen from various sites. The best methods are those that are specific, sensitive, fast, simple, reproducible, and low cost. In addition, they must provide information about the pathogenic potential of strains. By applying these methods, it has been possible to detect various foods involved in outbreaks of listeriosis and allow proper control by food inspection agencies and the food industry. These methods can be useful for monitoring critical control points in the food-handling environment to provide subsidies to apply corrective measures regarding cleaning and sanitization procedures to eliminate L. monocytogenes in the environment.
Footnotes
Acknowledgments
The authors thank Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG).
Disclosure Statement
No competing financial interests exist.