
Abstract
The contamination of oysters with human noroviruses poses a human health risk, since oysters are often consumed raw. In this study, human norovirus genogroup II was allowed to bio-accumulate in oysters, and then the effect of high-pressure processing (HPP) on human noroviruses in oysters was determined through a polymerase chain reaction (PCR)-based method with enzymatic pretreatment to distinguish infectious noroviruses. As a result, oysters could be artificially contaminated to a detectable level of norovirus genome by the reverse transcription-PCR. Concentrations of norovirus genome in laboratory-contaminated oysters were log normally distributed, as determined by the real-time PCR, suggesting that artificial contamination by bio-accumulation was successful. In two independent HPP trials, a 1.87 log10 and 1.99 log10 reduction of norovirus GII.17 genome concentration was observed after HPP at 400 MPa for 5 min at 25°C. These data suggest that HPP is a promising process of inactivation of infectious human noroviruses in oysters. To our knowledge, this is the first report to investigate the effect of HPP on laboratory-contaminated noroviruses in oysters.
Introduction
H
The shellfish industry has introduced high-pressure processing (HPP) to facilitate the shucking process. This procedure has been shown to dramatically improve the appearance, storability, texture, flavor, and yield of treated oysters (He et al., 2002; Murchie et al., 2005). Beyond cooking, HPP has been identified as a promising intervention to inactivate Vibrio spp. (Ye et al., 2012), and it is currently used commercially to eliminate a number of live Vibrio spp. in U.S. Gulf Coast oysters (Berlin et al., 1999; Cook, 2003). Furthermore, HPP has recently been suggested to be a promising intervention strategy to inactivate food-borne viruses in bivalve shellfish (Kingsley, 2014). When oyster homogenate, contaminated by inoculation of genogroup II.4 (GII.4), was treated at 300 MPa for 5 min at 6°C, a 3.51 log10 reduction of norovirus genome was observed (Ye et al., 2014). When oysters contaminated by an injection of GI.1 into the digestive tract were treated at 600 MPa for 5 min at 6°C, no norovirus infection was observed in clinical trials (Leon et al., 2011). However, a direct assessment of HPP on bio-accumulated human noroviruses within oysters is still unknown in terms of the evaluation of infectivity.
Due to the lack of a routinely available in vitro cell culture system or a small-animal model, reverse transcription-polymerase chain reaction (RT-PCR) is the most common detection method for noroviruses. Though this method shows advantages of rapidity and high sensitivity, it simply detects the presence of norovirus genome RNA and is unable to distinguish infectious from noninfectious virus (Baert et al., 2008; Lowther et al., 2010). Thus, the health risk of oysters may be overestimated by detecting virus genome derived from noninfectious viral particles (Uema et al., 2016). To avoid such false positives, several researchers have proposed enzymatic pretreatment of the sample by RNase before RT reaction to digest viral RNA-derived damaged particles (Nuanualsuwan and Cliver, 2002; Baert et al., 2008; Lamhoujeb et al., 2008); this has already been applied to the inactivation evaluation when studying norovirus responses to various interventions (Baert et al., 2008; Topping et al., 2009; Ye et al., 2014).
In this study, oysters were contaminated in water tanks with human noroviruses by bio-accumulation, and then the effect of HPP on human noroviruses in oysters was determined by using the PCR-based method with enzymatic pretreatment to distinguish infectious noroviruses from noninfectious ones.
Material and Methods
Oysters and test groups
Oysters (Crossostrea gigas) were purchased from Momonoura producer of the oyster consolidated company at Miyagi Prefecture in Japan from August 2016 to September 2016.
Preparation of the human norovirus challenge
The two different norovirus GII strains used in this study were purified from a stool specimen of patients with acute infectious gastroenteritis in Miyagi Prefecture, Japan. About 20 g of stool was suspended in 200 mL of distilled water and centrifuged at 10,000 g for 30 min. The supernatant, including norovirus particles, was collected and stored at −4°C. The genotype of norovirus GII in the specimen was found to be norovirus GII genotype 4 Sydney (GII.4 Sydney) and GII.17, which was revealed by RT-PCR and sequencing of the norovirus GII gene as previously described (Ueki et al., 2007).
Artificial contamination of oysters with norovirus
Artificial contamination of oysters with norovirus GII was conducted by bio-accumulation at Miyagi Prefecture Fisheries Technology Institute in accordance with our previous report (Ueki et al., 2007). Two independent water tanks were prepared, each containing 60 oysters in 150 L for the first batch, and 200 L for the second and third batches of sand-filtered seawater. The temperature of the water was set at 25°C ± 2°C, which corresponds approximately to the average water temperature of the oyster cultivation area in August and September 2016 in Miyagi Prefecture. Phytoplankton (Chaetoceros glacilis) was added to all the water tanks as feed for oyster cultivation. The virus was then added to one of the two water tanks, and the water was aerated at a rate of 4 L of air/min for 1 h. The contamination of oysters with virus was performed for 72 h under aeration, but contaminated oysters were transferred every 24 h to new water tanks in which test virus and phytoplankton were freshly prepared. Concentrations of noroviruses used for laboratory contamination are described in Table 1. The resulting 60 oysters were used as laboratory-contaminated oysters. Another 60 oysters, in the water tanks without virus, were used as native noncontaminated oysters.
The average number was calculated by dividing the sum on the number of positive oysters only.
The contamination of oysters with virus was performed for 72 h under aeration condition, and contaminated oysters were transferred every 24 h to water tanks in which freshly prepared test virus and phytoplankton were added. Genotype of test virus is GII.4 Sydney for the 1st batch, and GII.17 for both second and third batches. Concentrations (copy/L) of test virus in the tank were 1.08 × 106, 4.10 × 106, and 1.80 × 106 for the first, second, and third tanks, respectively.
Five out of 30 oysters died during 72 h of laboratory contamination.
Significant difference of detection rate observed between groups 3 and 4 (p < 0.001).
Significant difference of average genome copy number/g of digestive diverticula observed between groups 3 and 4 (p < 0.001).
HPP, high-pressure processing.
HPP of oysters
HPP was conducted at Momonoura producer of the oyster consolidated company at the Miyagi Prefecture. Thirty out of the 60 oysters from both native and laboratory-contaminated tanks were pressurized at 400 MPa for 5 min at 25°C. Consequently, a total of four groups were formed in the study (group 1: 30 native oysters; group 2: 30 native oysters with HPP; group 3: 30 laboratory-contaminated oysters; group 4: 30 laboratory-contaminated oysters with HPP). The experiment was repeated thrice. To avoid cross-contamination of norovirus between groups, each group of oysters was heat-sealed by using the Impulse food sealer as per the manufacturer's instructions. Oysters were refrigerated and shipped overnight to the Incorporated Foundation Kenbikyo-in for norovirus detection and quantification.
Sample preparation of viral suspensions and enzymatic pretreatment
For the preparation of viral suspensions, oysters were shucked, and the digestive diverticula were collected by dissection on the day of arrival to the laboratory. The sample was homogenized in nine times its volume of phosphate-buffered saline (PBS) solution (without magnesium and calcium). The homogenates were incubated with α-amylase (Wako) at a final concentration of 2.5 mg/mL for 1 h at 37°C and vortexed every 15 min. To concentrate the viral particles derived from digestive diverticula, the method provided by the Japanese Committee for Standardization Virus of Detection in Food (2010) was used. After centrifugation at 8000 g for 20 min, the supernatant was recovered. To concentrate the virus, polyethylene glycol 6000 (Sigma-Aldrich) and sodium chloride (Wako) were added at a final concentration of 12% and 5.8%, respectively. Eighteen hours after incubation at 4°C, the samples were centrifuged at 8000 g for 20 min. After removing the supernatant, the pellet was re-suspended in 400 μL of PBS (containing 0.5% Zwittergent 3–14 Detergent) (Merk). Fourteen units (1.4 μL) of RNase ONE™ ribonuclease (Promega), 10 μL of × 10 reaction mixture, and 18.6 μL of the distilled water were added to 70 μL of the resulting virus suspension, and the mixture (total volume of 100 μL) was incubated at 37°C for 1 h (Topping et al., 2009). The resulting enzyme-treated virus suspension was added to 100 μL of PBS(−) and then used for RNA extraction.
RNA extraction and RT
Viral RNA was extracted from 200 μL of viral suspension by using a High Pure Viral RNA kit (Roche Diagnostics) with recombinant DNase I (Roche Diagnostics) according to the manufacturer's instructions with the following modification. To promote recovery of RNA, 8 μL of MS2 RNA (Roche Diagnostics) was added to 400 μL of the binding buffer. cDNA synthesis of the first strand was performed by using High-Capacity cDNA Reverse Transcription kit (Life Technologies). To avoid detection of reverse transcripts due to fragmented genome (and thus noninfectious), Oligo dT primer was used for RT.
Detection of the capsid N/S region of VP1 gene
To amplify the partial capsid N/S region of noroviruses by RT-PCR, primers were prepared in accordance with previous reports (Imamura et al., 2016). An Applied Biosystems Veriti 96 Well Thermal Cycler (Applied Biosystem, CA, USA) was used, and the PCR protocol was as follows: an initial incubation for 15 min at 95°C, followed by 40 cycles of 94°C for 60 s, 50°C for 60 s, and 72°C for 2 min, with an additional 15 min elongation step at 72°C after the last cycle. This PCR procedure was repeated by using internal primers for nested PCR. Aliquots (8 μL) of the PCR products were analyzed by 2% agarose gel electrophoresis and 1 μg/mL ethidium bromide.
Quantification of norovirus GII by real-time PCR
Samples that were positive for the partial capsid N/S region of noroviruses by RT-PCR were processed by using real-time PCR to quantify norovirus capsid genes by using TaKaRa qPCR Norovirus (GI/GII) Typing Kit (Takara). The real-time PCR protocol included incubation for 10 s at 95°C, 45 cycles at 95°C for 5 s, and at 56°C for 34 s. All amplification was performed in duplicate. In case the R 2 value was more than 0.990, the real-time PCR procedure was regarded as successful, and the samples were then analyzed. The genome copy number was expressed as the genome copy number per one gram of digestive diverticula.
Statistical analysis
Significant differences in the detection rate and proportion of genotypes were determined by using the Fisher's exact test and the Mann–Whitney U test, respectively. Significant differences were defined as p < 0.01.
Results
Detection and quantification of norovirus GII in native/laboratory-contaminated oysters with/without HPP
Norovirus GI was not detected in all samples. Table 1 details the detection rate of norovirus GII in oysters in each tested group. Norovirus GII was detected in all laboratory-contaminated oysters, whereas it was not detected in any native oyster except for one sample in the third batch. Significant differences (p < 0.001) in detection rate were observed between groups 3 and 4 in the first batch. There were no significant differences in the detection rate between groups 3 and 4 in the second and third batches.
Average genome copy numbers per one gram of digestive diverticula in laboratory-contaminated oysters (group 3) in the first, second, and third batches were 5.89 × 105, 3.11 × 106, and 3.85 × 106, respectively. Average genome copy numbers per one gram of digestive diverticula in laboratory-contaminated oysters (group 4) in the second and third batches were 4.15 × 104 and 3.89 × 106, respectively. A 1.87 log10 and 1.99 log10 reduction was observed after HPP in the second and third batches, respectively. There was a significant difference in genome copy number between groups 3 and 4 in both the second and third batches (p < 0.01). Histograms demonstrating the distribution of genome copy number per one gram of digestive diverticula in laboratory-contaminated oysters with/without HPP were prepared (Fig. 1). Distribution was shifted to the left after HPP.
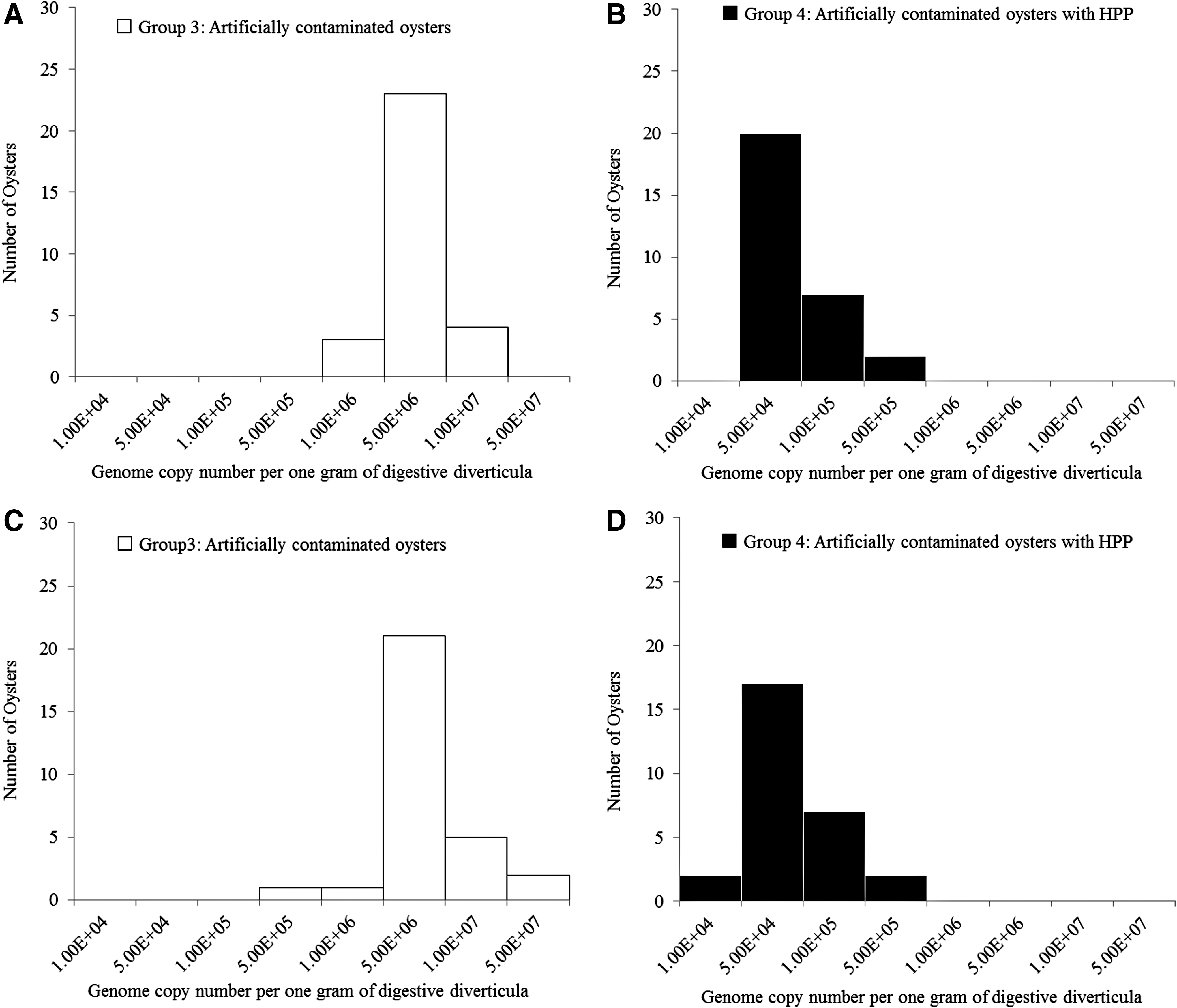
Histograms showing the distribution of genome copy number per one gram of digestive diverticula in laboratory-contaminated oysters with/without HPP.
Discussion
It is worthwhile to investigate the effect of HPP on noroviruses in oysters that bio-accumulated under similar conditions to the aqua-cultural environment. In this study, the contamination of oysters with noroviruses was conducted based on the method reported by Ueki et al. (2007). Consequently, it was possible to contaminate all oysters up to the level whereby the norovirus genome could be detected by RT-PCR, and quantified concentrations of norovirus genome were distributed normally, suggesting that this method appears to be effective in the preparation of oyster samples to evaluate procedures for efficient removal or inactivation of the viruses in oysters. However, norovirus concentrations in the laboratory-contaminated oysters were extremely high compared with those in naturally contaminated oysters. Naturally contaminated market oysters would not be as high in norovirus, and, therefore, an intervention that kills 2 logs of virus would likely have a strong impact on public health. Regarding the condition of oysters, artificial contamination should be avoided in the summer season, since mass mortality events are often observed in cultured oysters during the period after spawning in the summer season (Berthelin et al., 2000; Cheney et al., 2000; Huvet et al., 2004; Garnier et al., 2007). In the present study, oysters used for the first batch were collected on August 29, which corresponds approximately to the period just after spawning. We speculated that this was the reason that 10 out of 60 oysters died in the water tanks during the 72 h of artificial contamination. Thus, the data obtained from the first batch are considered unreliable data, since the conditions of oysters after spawning are unusual.
Recently, strain-dependent bio-accumulation in oysters was reported by Maalouf et al. (2011). They demonstrated that bio-accumulation of GI.1 occurred specifically in digestive tissues in a dose-dependent manner, and efficiency was parallel with ligand expression in the oyster, which was at its highest during cold months. In comparison, GII.4 displayed poor bio-accumulation, and it was recovered in almost all tissues without seasonal influence (Maalouf et al., 2011). In a previous report, we examined oysters collected between January and March in 2015, and we demonstrated that GII.17 was detected in 77 out of 89 of the oyster samples that were positive for GII (Imamura et al., 2016), indicating a strain-dependent affinity with oysters. Thus, both water temperature and norovirus genotypes may affect the levels of artificial contamination of oysters by noroviruses.
As there was no existing study investigating the efficacy of HPP when used against naturally present noroviruses in oysters, we decided to use experimental conditions based on a report demonstrating the effect of HPP on oyster homogenate inoculated with human noroviruses (Ye et al., 2014), so that noroviruses in the oysters would be inactivated. For this reason, the effect of HPP on laboratory-contaminated oysters was initially evaluated at 5 min, 400 MPa, and 25°C, which corresponds to the temperature of seawater in aqua-cultured areas, and this resulted in a logarithmic reduction in the genome concentration of norovirus. Though norovirus was detected even after HPP, it is notable that the levels of contamination in oysters prepared for the present study were notably higher than those of oysters collected from commercial harvest areas. We previously surveyed the noroviruses in oysters obtained from fish producers at six different sites (sites A, B, C, D, E, and F) in Japan once a month from October 2015 to February 2016, and the range of copy/g in GII-positive samples was found to be from 44 to 1534 copy/g (unpublished data). Thus, it is expected that the levels of noroviruses in commercial oysters may be reduced by HPP to levels that might not cause oyster-related gastroenteritis. In addition, the goal is to reach a contamination level of less than 1000 copies/g, since Lowther et al. (2012) reported that the geometric mean of the levels in samples obtained during the outbreak period (1048 copies/g) was higher by almost one order of magnitude than that in samples not obtained during the outbreak period (121 copies/g).
However, the effect of HPP on viruses is reported to be varied depending on the level of pressure, retention time, temperature, target virus (serotype/genotype), and sample environment in viruses (Noda, 2017). In addition, the application of lower pressure will be preferable to extend shelf life, and the oyster industry uses HPP at pressures less than 300 MPa for oyster shucking. It is well known that human noroviruses and other caliciviruses are highly sensitive to HPP at cooler pressurization temperatures (Kingsley, 2013). Thus, HPP at refrigeration temperatures needs to be considered to determine the efficacy of HPP on laboratory-contaminated oysters. Further studies are needed to determine the optimum conditions of processing oysters at as low a pressure as possible. Meanwhile, Kingsley (2014) demonstrated that higher pressures can be used as a food-borne virus intervention for human noroviruses and hepatitis A virus, without impacting consumer acceptance or the commercial viability of raw HPP-treated oysters.
For the detection of infectious noroviruses, Nuanualsuwan and Cliver (2002) reported that simultaneous treatment with protease K (PK) and RNase before conventional RT-PCR is useful. A recent study considering the necessity of PK treatment conducted by Noda et al. (2014) demonstrated that no significant difference was observed between simultaneous treatment of PK with RNase and RNase alone. Thus, we used RNase alone for pretreatment during sample preparation. Though this method has advantages in the detection of infectious norovirues, Baert et al. (2008) demonstrated that there is no correlation between the number of infectious particles and viral genomes after heat treatment regardless of the presence or nature of enzyme pretreatment. Thus, it is important for further elaboration of the detection method to investigate the infectivity of norovirus in the samples. However, pretreatment by RNase is considered logically reasonable, and it has a certain advantage in that it digests the virus genome derived from noninfectious virus. The culture system appears to be an ideal method for the assessment of infectious noroviruses, since the actual reduction might be underestimated by RT-PCR. In this regard, murine norovirus, a propagable norovirus, seems to have an advantage of evaluating susceptibility to HPP (Kingsley et al., 2007). Furthermore, replication of human noroviruses in stem cell-derived human enteroids reported by Ettayebi et al. (2016) will hopefully be applied widely in this field.
Conclusions
To our knowledge, this is the first report to investigate the effect of HPP on noroviruses that were bio-accumulated from contaminated water by oysters. Norovirus kinetics in laboratory-contaminated oysters in the present study appears to be very similar to that in naturally contaminated oysters. However, the effect of HPP on naturally present noroviruses in oysters is yet to be understood.
Footnotes
Acknowledgments
This study was performed as part of the surveillance/monitoring program of microbiological hazards of the Ministry of Agriculture, Forestry and Fisheries, Tokyo, Japan.
Disclosure Statement
No competing financial interests exist.