
Abstract
Rapid detection and timely treatment of diseases caused by foodborne pathogens is important for improving the curative efficiency and preventing the spread of disease. In this study, we developed an assay utilizing a recently introduced ultra-fast convection polymerase chain reaction (cPCR) in conjunction with a simple nucleic acid lateral flow (NALF) immunoassay for ultra-fast on-site molecular detection of foodborne pathogens. Two Salmonella enterica serovars, Salmonella Enteritidis and Salmonella Typhimurium, and Escherichia coli O157:H7 were used as the target pathogens. We confirmed the specific amplification of the target species with specifically designed modified primer sets for cPCR in singleplex and duplex modes. After cPCR amplification, we compared the detection specificity and sensitivity using agarose gel electrophoresis and NALF assays with one or two test lines. The cPCR amplicons were readily and sensitively detected using the NALF assay, and the sensitivity was comparable with that of agarose gel electrophoresis. To confirm the application of the assay in real-life samples, the assay was used to test artificially contaminated milk. Without sample pre-enrichment, the limit of detection (LOD) was 4.5 × 103 colony-forming units (CFU)/mL for Salmonella species; 4.5 × 104 CFU/mL to differentiate Salmonella Enteritidis and Salmonella Typhimurium; and 2.3 × 103 CFU/mL for E. coli O157:H7, in a duplex assay. With a 6 h pre-enrichment, the LOD was 4.5 CFU/mL for Salmonella Enteritidis and Salmonella Typhimurium, and 2.3 CFU/mL for E. coli O157:H7. The cPCR amplification took only 14 min, and the NALF assay took ca. 5 min. The total analysis time was less than 20 min. Based on these observations, we propose that the developed assay is simple, ultra-fast, and applicable for on-site detection of foodborne pathogens.
Introduction
M
To better control microbial contamination of food and, thus, to reduce foodborne illness, rapid and accurate pathogen detection methods are required for efficient monitoring of microbial pathogens. Traditional methods, such as immunological or biochemical identification, are time-consuming and laborious, which negatively impacts their applicability. To overcome these issues, various molecular approaches have been devised. Among the molecular assays, rapid detection using polymerase chain reaction (PCR) draws the most attention because of the speed and higher accuracy than the traditional serology-based microbial serotyping (Suo et al., 2010; Liu et al., 2012; Kamphee et al., 2015; He et al., 2016).
Convection PCR (cPCR) is a new technology (Hwang et al., 2009; Hwang, 2011). There, PCR involves the use of three heating plates for denaturation, annealing, and polymerization to generate convection in the PCR tubes. This method does not require ramping between temperatures employed in conventional thermocyclers, and thus the reaction time is dramatically reduced. The nucleic acid lateral flow (NALF) assay has been recently developed for the detection of foodborne pathogens, and pathogens and gene mutations that underlie human diseases (Kamphee et al., 2015; Shan et al., 2015; Singh et al., 2015; Wang et al., 2016; Zhang et al., 2017). An NALF assay device allows an indirect detection of amplified PCR products through the use of antibodies against specific primer tags, such as carboxyfluorescein (FAM), digoxigenin (DIG), and biotin. The aim of this study was to develop an ultra-fast and sensitive method for on-site detection of Salmonella spp. and E. coli O157:H7 by combining cPCR and the NALF immunoassay.
Materials and Methods
Bacterial strains and growth conditions
The bacterial strains used in this study were Salmonella Enteritidis, Salmonella Typhimurium (NCCP 14771 and NCCP 12219), and E. coli O157:H7 (NCCP 15739) from the National Culture Collection for Pathogens (NCCP) at the Korea Center for Disease Control & Prevention (Cheongju, Korea). Bacteria were handled as instructed by the NCCP. They were streaked on the Luria-Bertani (LB) agar (Duchefa, Haarlem, Netherlands) and incubated overnight at 37°C. For DNA purification, the bacteria were grown overnight in LB broth (Duchefa) at 37°C with shaking.
Preparation of genomic DNA
Genomic DNA (gDNA) from all strains was extracted using DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. DNA concentration was determined using a NanoDrop spectrophotometer (Thermo Scientific, Waltham, MA). Mixed gDNA samples were prepared by combining equal amounts of individually prepared gDNA. Any deviations from these procedures are stated in the text. gDNA copy numbers were calculated for 1 ng of DNA, based on the molecular weight of double-stranded DNA and chromosomal DNA size (
Primer design and cPCR
The primers used in this study are listed in Table 1. A specific Salmonella invasion protein A gene (invA) was used to design Salmonella-specific primers (SPP). The insertion element (IE; GenBank Accession No. Z83734) and a periplasmic protein gene (spy; GenBank Accession No. AE008757.1) were used to design serovar-specific primers for Salmonella Enteritidis and Salmonella Typhimurium, respectively. For E. coli O157:H7, the O157 antigen gene (rfbE; GenBank Accession No. S83460.1) and the H7 flagellar antigen gene (filC7; GenBank Accession No. AF228487) were used.
The cPCR mixture (20 μL) contained 1 × PalmTaq HS buffer (supplemented with 1.5 mM MgCl2), 0.2 mM dNTPs, 0.4 U PalmTaq high-speed DNA polymerase (Ahram Biosystems, Seoul, Korea), and primers for single or multiple PCR detection. For Salmonella detection using single-line NALF (NALF1), 10 μM SPP-FAM primers were used (singleplex detection). To differentiate between Salmonella Enteritidis and Salmonella Typhimurium using double-line NALF (NALF2), 10 μM ST-DIG primers and/or 8 μM SE-FAM primers were used (duplex detection). Generally, 1.6 ng (3 × 105 copies) of Salmonella gDNA was used as a template. For E. coli O157:H7 and NALF2, 10 μM each of O157-DIG and H7-FAM primers were used. Typically, 1.8 ng (3 × 105 copies) of E. coli O157:H7 gDNA was used as a template. PCR was performed using a battery-operated convection thermal cycler Palm PCR device (G2-12; Ahram Biosystems). The speed was set to T1, and the annealing temperature was set to 56°C. PCR reactions involved 20 cycles (14 min), unless stated otherwise. All experiments were performed at least in triplicate.
Detection of PCR amplification products by electrophoresis and NALF analysis
Upon completion of the cPCR, an aliquot of the PCR mixture was analyzed by 1.5% agarose gel electrophoresis (25 min at 135 V). PCR products were visualized with ethidium bromide staining and an imaging system (Ultra-Lum, Carson, CA).
The NALF cassette (Ahram Biosystems) was designed to indirectly detect amplified PCR products using antibodies against primer tags, such as FAM, DIG, and biotin. Gold-colloidal particles were conjugated with the antibodies to produce a colored product when the antibody bound its specific antigen. Briefly, the NALF assay cassette consisted of a sample application pad, a conjugation pad, nitrocellulose, and an absorption pad. When the NALF assay was used to detect DNA amplification, 2 μL of PCR product was diluted with 100 μL of the NALF buffer. The diluted solution was added to the reservoir of the NALF cassette, and waited for until the lateral flow was completed. The conjugation pad contained gold nanoparticles conjugated with anti-biotin antibodies (gold-anti-biotin complex). The flow direction was from the conjugation pad to the absorption pad. DNA amplicons bound to the gold-anti-biotin complex migrated to the test lines (T, or T1 and T2). Anti-FAM antibody was deposited at the test lines T and T1; and anti-DIG antibody was deposited at test line T2. The control line was coated with a goat anti-mouse antibody. Typically, the reaction development took ca. 5 min. The assay outcomes were documented by photographing. Antibodies used in NALF were purchased from Jackson ImmunoResearch Laboratoires (West Grove).
Preparation of artificially contaminated milk and PCR reaction
Fresh milk was purchased from the local market. For the experiment, 1 mL of milk was inoculated with 0.1 mL of different amounts of viable microbial cells (Salmonella Enteritidis or Salmonella Typhimurium, 4.5 × 104–4.5 × 100 colony-forming units (CFU)/mL; E. coli O157:H7, 2.3 × 104–2.3 × 100 CFU/mL). The milk was diluted with 9 mL of buffered peptone water (Oxoid, United Kingdom) and the milk was assayed immediately after contamination or the milk was incubated at 37°C for 6 h in a shaking incubator (Vision Scientific, Buchun, Korea). Next, DNA was purified from 1 mL of the samples, as described above. Finally, 1 μL of the DNA eluate was used as a template, and cPCR and detection were performed as described above.
Results
Specific and sensitive detection of foodborne pathogens using a combined cPCR and NALF1 immunoassay
The cPCR was performed with the primers designed for the detection of any S. enterica serovars (SPP-FAM, Table 1). Strong DNA amplification signal was detected with gDNA from both Salmonella serovars, Salmonella Enteritidis and Salmonella Typhimurium, as expected, but not with the other member of the Enterobacteriaceae, E. coli O157:H7, by agarose gel analysis (Fig. 1A, upper panel). Aliquots of the PCR reactions were used for the NALF assay. As shown in the lower panel of Figure 1A, the same results were obtained with the NALF assay: the amplicons of gDNA from both Salmonella strains were clearly observed as red lines in the NALF cassette, but no detectable colored line was developed when aliquots of E. coli O157:H7 gDNA were used. The sensitivity of the NALF assay was similar to that of agarose gel analysis (Fig. 1B). Approximately 3 × 101 copies of the chromosome of Salmonella species were detectable by the cPCR/agarose gel analysis, and the same was observed for the cPCR/NALF assay. Similar results were obtained for Salmonella Typhimurium (Fig. 1C).
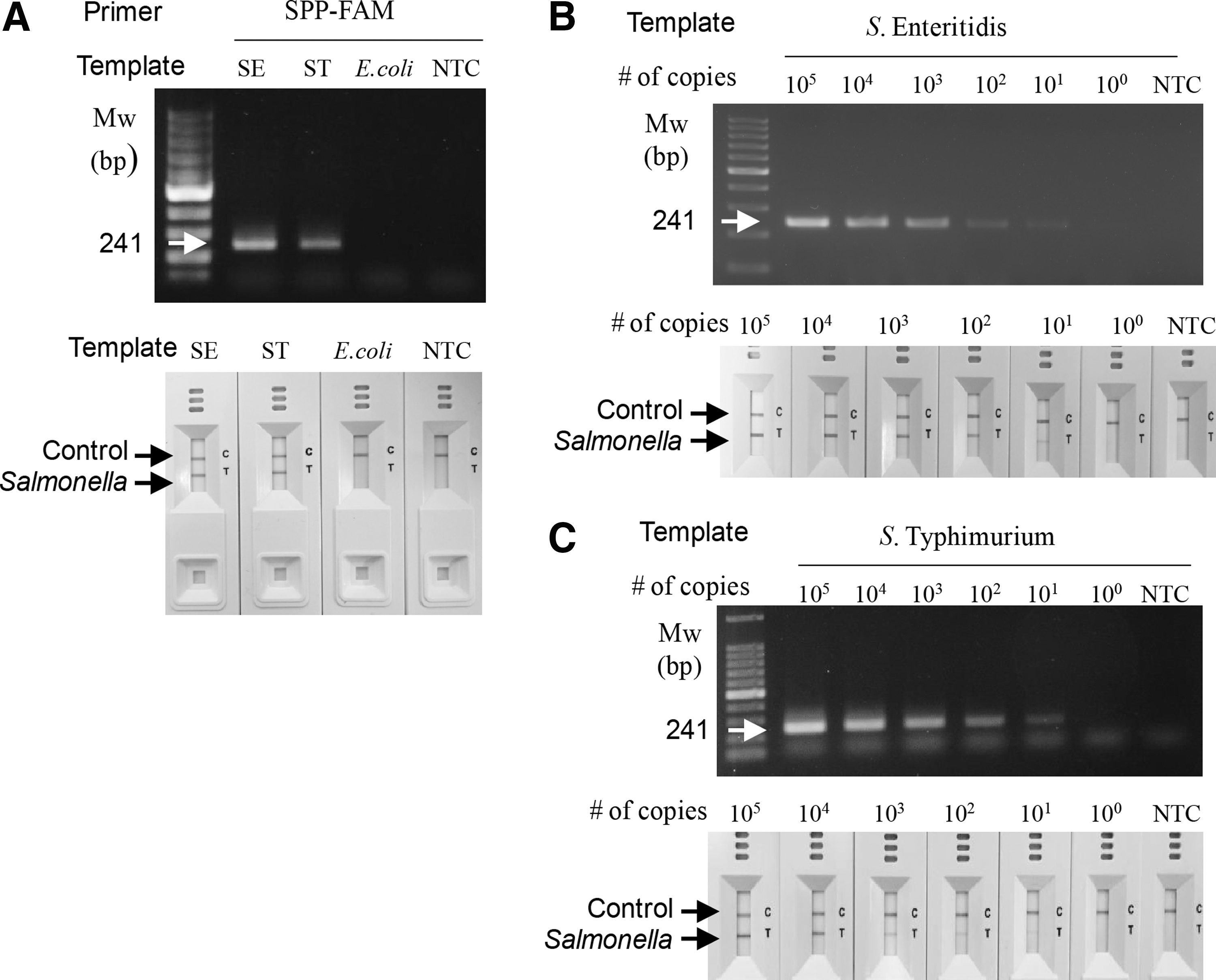
Specific and sensitive detection of Salmonella species using cPCR and the NALF1 assay. gDNA isolated from Salmonella Enteritidis (SE) and Salmonella Typhimurium (ST) were used as templates in the ultra-fast cPCR and NALF immunoassay with a single test line. DNA amplicons were analyzed by agarose gel electrophoresis or the NALF assay. The PCR operation time was 14 min (equivalent to 20 cycles).
Specific and sensitive detection of foodborne pathogens using a combined cPCR and NALF2 immunoassay
Next, we used the NALF2 assay to differentiate between the two S. enterica serovars, Salmonella Enteritidis and Salmonella Typhimurium. Following a cPCR reaction with primer sets SE-FAM and ST-DIG (Table 1), the amplicons were analyzed by both agarose gel electrophoresis and the NALF assay. Specific amplification of the Salmonella Enteritidis and Salmonella Typhimurium gDNA was clearly detected (Fig. 2A). The results of the NALF assay (lower panel of Fig. 2A) were congruent with those of electrophoresis (upper panel of Fig. 2A). The limit of detection (LOD) was as low as 3 × 102 copies of gDNA for Salmonella Enteritidis and Salmonella Typhimurium for both cPCR/agarose gel analysis and cPCR/NALF2 assay (Fig. 2B).
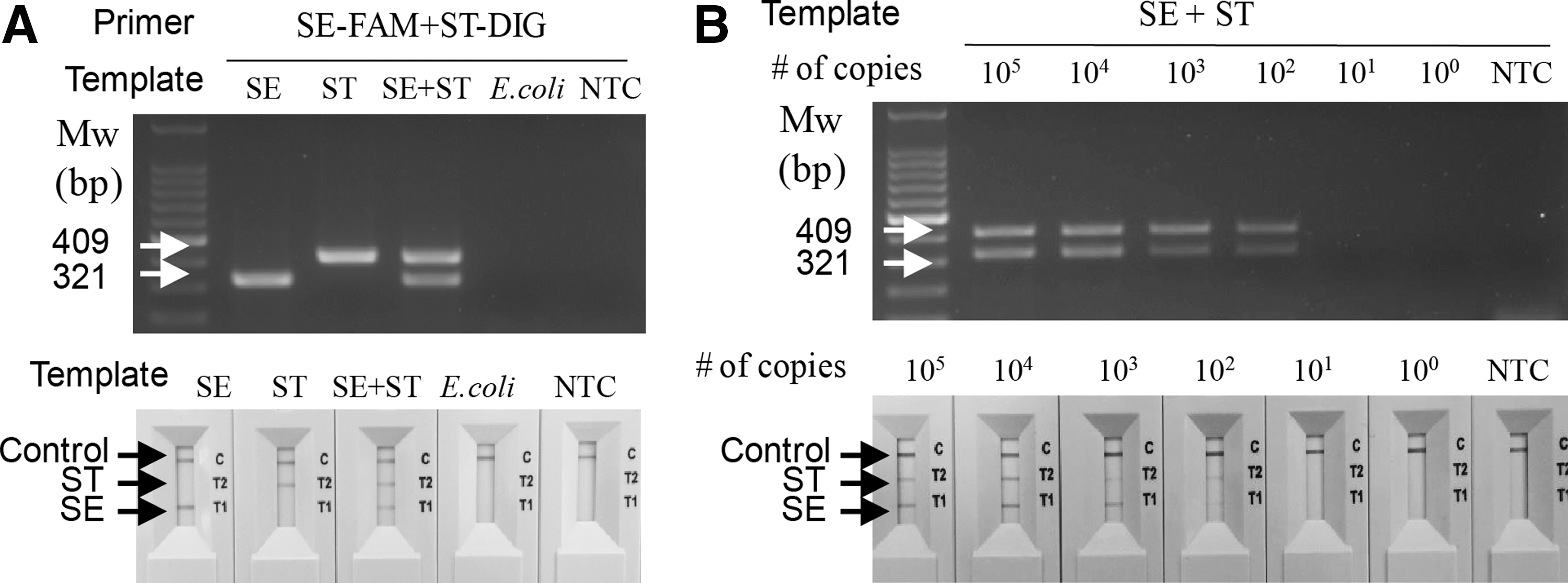
Specific and sensitive detection, and differentiation, of Salmonella Enteritidis and Salmonella Typhimurium using cPCR and NALF2 assay.
To extend the usage of the combination of cPCR and the NALF assay, we then attempted to detect E. coli O157:H7. We chose to target rfbE and filC7, which encode the O and H antigens, respectively (Fields et al., 1997; Maurer et al., 1999). As shown in Figure 3, 213 and 108 bp products were amplified from gDNA of E. coli O157:H7. No products were observed with gDNA of Salmonella Enteritidis or Salmonella Typhimurium (Fig. 3A). The LOD of the E. coli O157:H7 gDNA both in the cPCR/NALF assay and cPCR/agarose gel analysis was 3 × 102 copies (Fig. 3B).
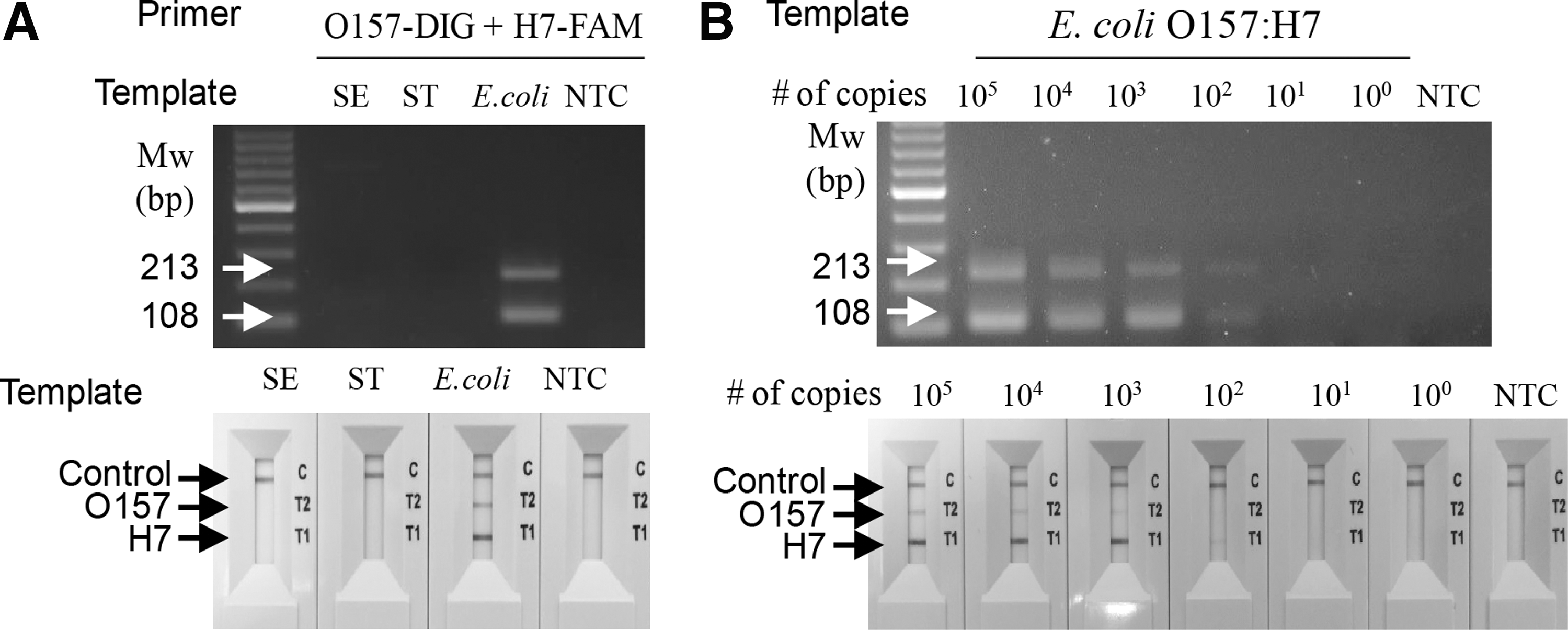
Specific and sensitive detection of E. coli O157:H7 using cPCR and the NALF2 assay.
Detection of foodborne pathogens in artificially contaminated milk without pre-enrichment
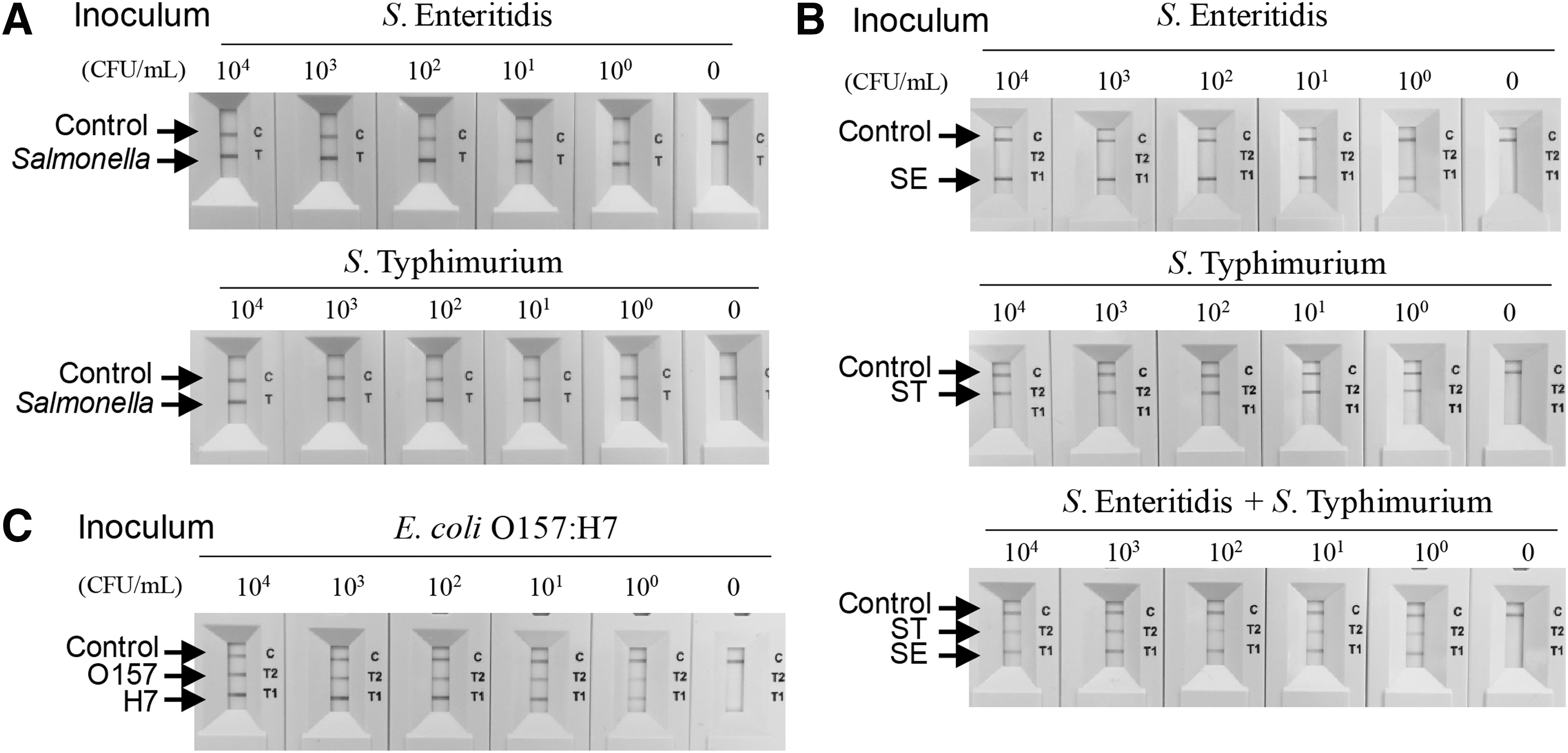
To validate the developed assay, we attempted to detect S. enterica serovars Enteritidis and Typhimurium in artificially contaminated milk. Fresh milk was inoculated with either Salmonella Enteritidis or Salmonella Typhimurium cells at 4.5 × 104–4.5 × 100 CFU/mL. Then, gDNA was purified and used in cPCR, without any pre-enrichment incubation step. As shown in Figure 4, LOD of Salmonella Enteritidis was ca. 4.5 × 103 CFU/mL when cPCR was combined with either electrophoresis or NALF1 assay (Fig. 4A, lower panel). No DNA product was detected on agarose gel or in the NALF cassette window when uninoculated samples were assayed. Similar results were obtained with milk artificially contaminated with Salmonella Typhimurium (Fig. 4B).

Detection of Salmonella Enteritidis and Salmonella Typhimurium in artificially contaminated milk, without pre-enrichment. Fresh milk was contaminated with 4.5 × 104–100 CFU/mL Salmonella Enteritidis
Next, milk was simultaneously inoculated with serially diluted Salmonella Enteritidis and Salmonella Typhimurium cultures, and gDNA was immediately isolated without any pre-enrichment step. Duplex cPCR was conducted (Fig. 4C). When the cPCR/electrophoresis combination was used, ca. 4.5 × 104 CFU/mL contamination by both Salmonella Enteritidis and Salmonella Typhimurium was detected. The same LOD values were obtained using the cPCR/NALF combination.
We then extended the assay to detecting E. coli O157:H7 in the artificially contaminated milk. gDNA was purified from milk contaminated with serially diluted E. coli O157:H7 culture, without pre-enrichment, and used as a template in cPCR. As shown in Figure 5, two amplified DNA bands were detected by electrophoresis, and the LOD was ca. 2.3 × 103 CFU/mL. As above, two red lines were observed in the NALF2 assay and the LOD was the same as that of the electrophoresis analysis. No DNA product was observed after the electrophoresis and the NALF assay when uninoculated samples were analyzed.
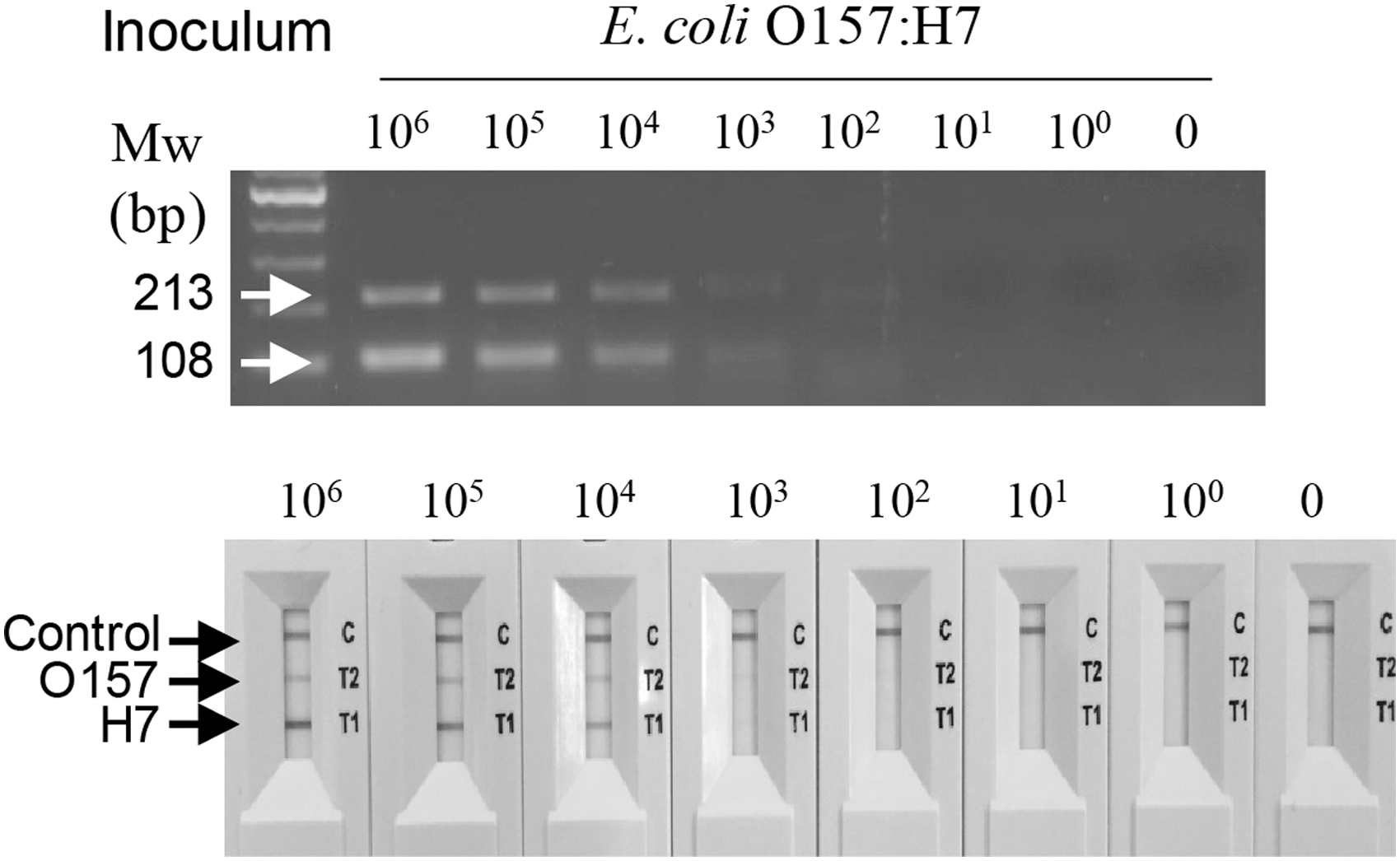
Detection of E. coli O157:H7 in artificially contaminated milk, without pre-enrichment. Fresh milk was contaminated with 4.5 × 104–100 CFU/mL E. coli O157:H7. gDNA was isolated without pre-enrichment and used as a template for cPCR. The resulting amplicons were analyzed by agarose gel electrophoresis and NALF2 assay. 0, uninoculated sample; CFU, colony-forming units; cPCR, convection polymerase chain reaction; gDNA, genomic DNA; Mw, molecular weight marker; NALF, nucleic acid lateral flow.
Detection of foodborne pathogens in artificially contaminated milk with a 6 h pre-enrichment step
Next, we pre-enriched milk contaminated with Salmonella Enteritidis, Salmonella Typhimurium, and E. coli O157:H7 for 6 h, following which gDNA was isolated and used as a template in cPCR. Agarose gel electrophoresis and the NALF assay were both used to analyze the cPCR results. The results of the NALF assay are shown in Figure 6. Using the NALF1 assay, specifically designed for the detection of any S. enterica serovar, both Salmonella Enteritidis and Salmonella Typhimurium were clearly detected, with the LOD of 4.5 CFU/mL (Fig. 6A). Using the NALF2 assay specific to individual Salmonella species, both Salmonella Enteritidis and Salmonella Typhimurium were detected, with the LOD of 4.5 CFU/mL (Fig. 6B). In another NALF2 assay specific to E. coli O157:H7, both O157 and H7 gene amplification products were detected, with the LOD of 2.3 CFU/mL (Fig. 6C).
Detection of foodborne pathogens in contaminated milk, with a 6 h pre-enrichment. Fresh milk was contaminated with 4.5 × 104–100 CFU/mL Salmonella Enteritidis or Salmonella Typhimurium
Discussion
In this study, we developed ultra-fast assays for on-site foodborne pathogen detection using cPCR and the NALF technologies. For an assay to be suitable for on-site use, it should be sufficiently rapid and sensitive, and not require complex equipment. Hence, a small battery-operated hand-held PCR device utilizing the recently developed cPCR technology is suitable for on-site use. Comparative data with the conventional PCR, specificity and sensitivity of cPCR technology with multiple strains of pathogens were demonstrated in previous studies (Kim et al., 2017; Song et al., 2017). However, amplicon detection by agarose gel electrophoresis is not easily performed on-site, where the electricity supply is limited. Hence, we combined cPCR and the NALF immunoassay. The NALF immunoassay is performed using a small device, the NALF cassette, which is easy to carry and handle. We used battery-operated homemade centrifuge for DNA preparation. Our data suggested that the NALF assay is a good alternative of gel electrophoresis, retaining its specificity and sensitivity. The cPCR amplification took 14 min, and the NALF assay took 5 min. Therefore, the detection of foodborne pathogens such as Salmonella species and E. coli O157:H7 using this assay was done within 20 min. Since DNA extraction takes ∼30 min, the whole experiments can be done within 50 min. The combination of the two technologies resulted in a simple, ultra-fast, and easy-to-use assay for on-site detection of foodborne pathogens.
The use of the NALF assay entails a few considerations. Since the color development in the assay depends on the antibody binding to an antigen coupled to the DNA amplicon but not on the specific amplification of target DNA, a possibility exists of nonspecific color development with nonspecifically amplified DNA. Therefore, primers used should be designed carefully so as to not generate nonspecific products during the PCR reactions. In addition, the crossover contamination between evaluated samples should be carefully monitored.
We used artificially contaminated milk as a model system to confirm that the developed assays could be used with real samples. Since the assays were to be used on-site, the artificially contaminated milk was initially not pre-enriched and gDNA was isolated immediately after bacterial inoculation. As evidenced, 2.3–4.5 × 103–104 CFU/mL pathogens could be detected within 14 min of cPCR amplification, and 5 min of NALF1 and NALF2 detection. When the artificially contaminated milk was pre-enriched for 6 h, the developed molecular assay detected as low as 2.3–4.5 CFU/mL S. enterica serovars Enteritidis and Typhimurium, and E. coli O157:H7 because the pathogens were proliferated during pre-enrichment.
The sensitivity of the assay was better than that of the previously described assays. Generally, sample pre-enrichment of 6–24 h was employed in other studies. The data presented here suggested that the assay developed herein was very sensitive and ultra-fast compared with other similar such assays. Typically, LOD of 107–108 CFU/mL for Salmonella without pre-enrichment is reported for the lateral flow immunological assay (Kuijpers et al., 2018). In other studies, LOD of 104–106 CFU/mL for Salmonella after a 24 h pre-enrichment (McCarthy et al., 2009; Moongkarndi et al., 2011); 2.3 CFU/mL for E. coli O157:H7 without pre-enrichment; and 2.3 × 103 CFU/mL for E. coli O157:H7 after an 18 h enrichment (Zhao et al., 2010) were reported for the lateral flow immunological assay. For molecular testings the LOD acquired was 101–103 CFU/mL, 103–104 CFU/mL, or 101–102 CFU/g without pre-enrichment (Zheng et al., 2014; Hanabara and Ueda, 2016; Miao et al., 2018). And the LOD of 101 CFU/g with 6 h pre-enrichment or 102–104 copies with 16–18 h incubation were observed in real-time PCR assays using real samples (Maurischat et al., 2015; Miao et al., 2018). These molecular testing results were obtained using real-time PCR assays with real samples. However, these real-time assays cannot be used for on-site detection of foodborne pathogens.
Conclusions
In this study, we developed an ultra-fast, specific, and sensitive molecular diagnostic assay for on-site detection of foodborne pathogens. The assay combined the recently developed cPCR and NALF methodologies. The reaction time of the combined cPCR/NALF assay was less than 20 min. The assay was substantially more rapid than other similar such assays. The LOD of the assay performed using artificially contaminated milk was ca. 4.5 × 103 CFU/mL for S. enterica serovars, 4.5 × 103 CFU/mL for a dual detection of Salmonella Enteritidis and Salmonella Typhimurium, and 2.3 × 103 CFU/mL for E. coli O157:H7, with no pre-enrichment. With a 6 h pre-enrichment step, the LOD was 4.5 CFU/mL for S. enterica serovars Enteritidis and Typhimurium, and 2.3 CFU/mL for E. coli O157:H7. We believe that the ultra-fast speed, specificity, sensitivity, and on-site usability of the molecular detection method presented in this study offer a reliable strategy for foodborne pathogen detection on-site. By designing additional specific primers, this method can easily be extended to on-site detection of other pathogens.
Footnotes
Acknowledgment
This work was supported by grant 10080151 from Korea Evaluation Institute of Industrial Technology, funded by the Ministry of Trade, Industry and Energy, Korea.
Disclosure Statement
No competing financial interests exist.