
Abstract
Increasing clinical significance of coagulase-negative staphylococci requires effective methods for species identification and genotyping. In this study, six housekeeping genes (femA, ftsZ, gap, pyrH, rpoB, and tuf) with extensive allelic polymorphisms were identified and evaluated to develop a comprehensive multilocus sequence typing (MLST) scheme. Selected primers were capable of amplification of the six loci from all of the 180 Staphylococcus strains belonging to 18 different species. Sequence analysis of each locus (44–63 alleles) revealed higher nucleotide diversity than 16S rRNA (28 alleles). Phylogenetic analysis of the concatenated sequences (3054 bp) of the six loci provided accurate species identification and highly discriminatory typing for all the strains. Multilocus allelic analysis of the 180 Staphylococcus strains generated 103 different sequence profiles, suggesting high genetic diversity of the strains. For example, 30 S. aureus, 37 S. epidermidis, 32 S. haemolyticus, and 14 S. hominis strains were typed into 15, 21, 11, and 10 sequence profiles, respectively. Compared with published MLST schemes that restrict on a few particular species, this new scheme both achieved similar discrimination for typing S. aureus, S. epidermidis, S. haemolyticus, and S. hominis and provided sufficient discriminatory power for typing additional opportunistic species, such as S. cohnii, S. capitis, and S. warneri. Importantly, the comprehensive MLST scheme for Staphylococcus strains provides a better genotyping tool for understanding the phylogeny of coagulase-positive Staphylococcus aureus strains.
Introduction
Staphylococcus is a widespread Gram-positive (GP) bacterium that comprises 52 species and 28 subspecies (
For decades, a number of studies that had been reported with regard to the genetic diversity of clinical staphylococcal isolates have primarily focused on S. aureus in China (Chen et al., 2014; Yang et al., 2018), whereas information on clonal diversity of CoNS strains is limited. Over the past years, several molecular typing methods, such as staphylococcal cassette chromosome mec (SCCmec) typing, multilocus sequence typing (MLST), spa typing, coa typing, random amplified polymorphic DNA analyses (RAPD), and enterobacterial repetitive intergenic consensus PCRs (ERIC–PCR), have been developed for the study of genetic diversity of staphylococcal isolates (Xu et al., 2008a, b; Song et al., 2015; Liu et al., 2016). Among them, SCCmec typing classifies Staphylococcus strains based on the diversity of a mobile genetic element named SCCmec, an important determinant of methicillin resistance. With 11 established SCCmec types and various subtypes, it has proven to be valuable for genotyping methicillin-resistant Staphylococcus strains (Liu et al., 2016). MLST has been commonly used for bacterial genotyping based on the nucleotide sequence of six or seven housekeeping genes, and the most important advantage is its capability to genotype a wide range of microorganisms, including both CoPS and CoNS species (Perez-Losada et al., 2013). Other genotyping methods, such as RAPD and ERIC-PCR, have shown poor discrimination power for CoNS strains.
MLST of Staphylococcus isolates in China is of particular interest due to the high occurrence and widespread distribution of the organism in different regions of the country. In Guangzhou, the representative of Southern China, ST239 was the only prevalent genotype of methicillin-resistant Staphylococcus aureus (MRSA) during 2001–2006 (Xu et al., 2011) and 2001–2012 (Deng et al., 2015). In Northern China, 87.1% of the isolated MRSA belonged to ST239-MRSA-III-spa t037 or ST239-MRSA-III-spa t030 (Chen et al., 2010), indicating that ST239 had been the only prevalent genotype of MRSA through mainland China for the first decade of 21st century. In addition, longitudinal studies have shown that ST5, ST8, ST59, ST188, and ST239 are the predominant S. aureus clonal lineages to cause human infections in China (Xie et al., 2011; Yang et al., 2018). The emergence of new types of strains poses a challenge to human health and demands effective detection and control strategies.
Although MLST has been applied to discriminate a number of Staphylococcus strains, currently available schemes are limited to only six species of S. aureus (Enright et al., 2000), S. epidermidis (Thomas et al., 2007), S. haemolyticus (Cavanagh et al., 2012), S. hominis (Zhang et al., 2013), S. lugdunensis (Chassain et al., 2012), and S. pseudintermedius (Solyman et al., 2013). Furthermore, the loci of housekeeping genes used in MLST vary in different species. Emergence of new staphylococcal species requires the development of new MLST schemes for genotyping and evolutionary analysis of the organisms. To better understand the genetic diversity and population structures of Staphylococcus strains belonging to different species, this study aims to develop a comprehensive MLST scheme with high discriminatory power for simultaneous identification and genotyping of the pathogens.
Materials and Methods
Bacterial strains
A total of 180 Staphylococcus strains of 18 species, isolated from various clinical specimens, food and drug samples, and industrial environmental samples in China between 2012 and 2015, were used in this study. This collection consists of 30 S. aureus, 37 S. epidermidis, 32 S. haemolyticus, 21 S. cohnii, 14 S. hominis, 9 S. capitis, 9 S. warneri, 4 S. pasteuri, 4 S. pseudintermedius, 4 S. simulans, 4 S. xylosus, 3 S. saprophyticus, 2 S. sciuri, 2 S. lentus, 2 S. gallinarum, 1 S. lugdunensis, 1 S. arlettae, and 1 S. hyicus. In addition, American Type Culture Collection (ATCC) strains ATCC13565 (S. aureus), ATCC12228 (S. epidermidis), ATCC29062 (S. simulans), ATCC51129 (S. pasteuri), ATCC29663 (S. pseudintermedius), ATCC27844 (S. hominis), ATCC27836 (S. warneri), and ATCC35539 (S. gallinarum), and China Center of Industrial Culture Collection (CICC) strains CICC10897 (S. lentus), CICC21602 (S. lentus), CICC21723 (S. capitis), CICC21722 (S. capitis), CICC10290 (S. capitis), CICC10499 (S. pseudintermedius), CICC24064 (S. simulans), CICC22122 (S. xylosus), and CICC22941 (S. saprophyticus) were included as reference strains. All the strains were purified from single colonies on tryptic soy agar plates. Genomic DNA of the strains was extracted from overnight cultures grown at 37°C in tryptic soy broth using a DNeasy Kit (TAKARA BIO, INC., Dalian, China).
Phenotypic and genotypic identification of Staphylococcus strains
Phenotypic identification of the Staphylococcus strains was carried out by Gram staining and colony morphology, and then confirmed by VITEK 2 GP identification cards (BioMerieux, Inc., Marcy l'Etoile, France). Samples with an identity score <90% were considered insufficient for a positive result. Genotypic identification was performed by PCR amplification and sequencing of 16S rRNA as previously described (Becker et al., 2004). Bidirectional DNA sequencing was performed for all of the PCR products by commercial company (Sangon Co., Ltd., Shanghai, China). The resulting sequence of 16S rRNA was subjected to a Basic Local Alignment Search Tool search. In compliance with the Clinical and Laboratory Standards Institute guidelines for the interpretation of 16S rRNA sequences, a query sequence with ≥98.65% identity to a reference in GenBank was acceptable for species identification (Kim et al., 2014).
Comprehensive MLST of Staphylococcus strains
Six housekeeping genes (femA, ftsZ, gap, pyrH, rpoB, and tuf) (Table 1) were selected for species identification and comprehensive MLST based on following criteria: (1) present in all the Staphylococcus strains, (2) requiring only one set of primers for PCR amplification and sequencing of an internal fragment, (3) present in a single copy in a bacterial genome, and (4) contributing to a high discriminatory power (Urwin and Maiden, 2003; Maiden, 2006; Didi et al., 2014). PCR primers designed to amplify the housekeeping genes (Table 1) were synthesized at Sangon Co., Ltd. PCR was performed on a GeneAmp PCR system 9700 thermal cycler (Applied Biosystems, Foster City, CA) in a final volume of 25 μL containing 0.50 μM each primer, 12.5 μL of Premix Taq (TAKARA BIO, INC.), and 2 μL of extracted DNA sample. The amplification conditions consisted of 95°C for 90 s, followed by 30 cycles of 20 s at 94°C, 20 s at 50°C, 40 s at 72°C, and a final extension at 72°C for 5 min. PCR products were resolved and visualized by electrophoresis in gels containing 1.2% agarose and 0.5 μg/mL ethidium bromide. After that, all the amplified DNA fragments were sequenced commercially in both directions (Sangon Co., Ltd.), and the sequence reads were analyzed using Lasergene 7.0 (DNAStar, Madison, WI).
Primers for Polymerase Chain Reaction Amplification of Housekeeping Genes in Staphylococci
The letter Y is nucleotide C or T; R is nucleotide A or G; M is nucleotide A or C; W is nucleotide A or T; B is nucleotide G, T, or C; D is nucleotide G, T, or A; H is nucleotide A, T, or C; and N is nucleotide A, G, C, or T.
MLST of S. aureus, S. epidermidis, S. haemolyticus, and S. hominis strains
MLST of 30 S. aureus strains was performed by amplifying and sequencing the internal DNA fragments of 7 housekeeping genes (arcC, aroE, glpF, gmk, pta, tpi, and yqiL) (Enright et al., 2000). Thirty-seven S. epidermidis strains were typed by sequencing 7 loci in arcC, aroE, gtr, mutS, pyrR, tpiA, and yqiL (Thomas et al., 2007). Thirty S. haemolyticus strains were typed using the MLST scheme by sequencing 7 loci in arcC, SH1200, hemH, leuB, SH1431, cfxE, and RiboseABC (Cavanagh et al., 2012). In addition, 14 S. hominis strains were typed using the MLST scheme based on 6 housekeeping genes (arcC, glpK, gtr, pta, tpiA, and tuf) (Zhang et al., 2013). Allelic profile of each strain was determined by analyzing the sequence data in the public MLST database (
Population genetic and phylogenetic analysis
Nucleotide sequences of each locus from 180 Staphylococcus strains were aligned using the MEGA 7.0 software. Phylogenetic trees were constructed from the sequence alignments using the neighbor-joining method and bootstrapping algorithm (1000 replicates) in the software (Kumar et al., 2016). DNA sequence polymorphism was analyzed using the DnaSP v5.10 software. The numbers of alleles, polymorphic sites, nucleotide diversity per site (π), and allelic diversity (HD) were computed for each locus of the strains (Librado and Rozas, 2009). Simpson's index of diversity (D), a discriminatory index of a microbial typing method, was calculated as described by Hunter and Gaston (Hunter and Gaston, 1988).
Nucleotide sequences
DNA sequences of the femA, ftsZ, gap, pyrH, rpoB, and tuf loci from 180 Staphylococcus strains have been deposited in GenBank under accession numbers MF041991 to MF042162 for femA, MF620132 to MF620303 for ftsZ, MF620304 to MF620475 for gap, MF620476 to MF620647 for pyrH, MF620648 to MF620819 for rpoB, and MF620820 to MF620991 for tuf.
Results
Phenotypic and genotypic identification of Staphylococcus strains
Phenotypic and genotypic tests of 180 Staphylococcus isolates were performed by using the VITEK 2 identification system and 16S rRNA sequencing, respectively. Our data showed that 16S rRNA sequence of S. capitis and S. caprae strains had 100% identity, resulting in S. capitis strains could not be distinguished by16S rRNA sequence analysis. Strains from other 17 different Staphylococcus species listed in Table 2 could be accurately identified by 16S rRNA sequence analysis, whereas by VITEK 2 system, four S. pasteuri strains were misidentified as S. warneri (probability >95%), indicating some phenotypic similarity between the S. pasteuri and S. warneri species. The phenotypic results of the remaining 176 strains were found to be consistent with the genotypic results.
Species Identification of Staphylococcus Isolates by 16S rRNA Sequencing and VITEK 2 System
The strains of S. capitis and S. caprae were not distinguished by 16S rRNA sequencing.
Four S. pasteuri strains were falsely identified as S. warneri (probability >95%) by VITEK 2 system.
Nucleotide sequence variation of each housekeeping locus
Six housekeeping genes (femA, ftsZ, gap, pyrH, rpoB, and tuf) were selected for the species identification and genotyping of Staphylococcus strains in this study. The internal DNA fragment of each housekeeping gene was successfully amplified and sequenced from all the 180 isolates. Nucleotide sequences of the fragments, ranging in size from 474 to 534 bp, were used for analyzing sequence diversity of each locus (Table 3). The number of alleles for each housekeeping gene ranged from 44 (tuf) to 63 (rpoB) among these strains. The number of polymorphic sites on a given locus varied from 144 (tuf) to 300 (femA). Nucleotide diversity (π), the average number of pairwise nucleotide differences per site, ranged from 0.061 (tuf) to 0.209 (femA). Nevertheless, the numbers of alleles (28) and polymorphic sites (67), and nucleotide diversity (0.019) for 16S rRNA are substantially low. Taken together, these results demonstrate that the nucleotide variation of six housekeeping genes is much greater than 16S rRNA, suggesting this new MLST scheme may have a high discriminatory power for the identification and genotyping of Staphylococcus strains.
Nucleotide Sequence Variation of Housekeeping Loci and 16S rRNA in 180 Staphylococcus Strains
Identification and genotyping of Staphylococcus strains by comprehensive MLST
A total of 3054 nucleotides across the 6 housekeeping gene loci (femA, ftsZ, gap, pyrH, rpoB, and tuf) were sequenced from each strain. The phylogenetic relationship of 180 Staphylococcus strains was generated from the concatenated sequences of the 6 loci (Fig. 1). The neighbor-joining tree shows that the strains in the same species are clustered together, indicating they are genetically closely related to each other. Furthermore, phylogenetic analysis of six housekeeping genes not only accurately identified all the Staphylococcus strains at species level (Fig. 1) but also provided sufficient discrimination for genotyping different staphylococcal species (Table 4). Multilocus allelic analysis of the 6 housekeeping genes shows 103 distinct sequence profiles in 180 Staphylococcus strains of 18 species (Table 4). For example, 30 S. aureus strains were assigned to 15 sequence profiles, 37 S. epidermidis strains to 21 sequence profiles, and 32 S. haemolyticus strains to 11 sequence profiles. Furthermore, Simpson's index of diversity (D) was calculated for some species (number of strains >10). Based on the numbers of Simpson's index, the comprehensive MLST scheme provided a high discriminatory power for typing S. epidermidis (D = 0.955) and S. aureus (D = 0.929) strains, but relatively weak discriminatory power for S. haemolyticus strains (D = 0.716).
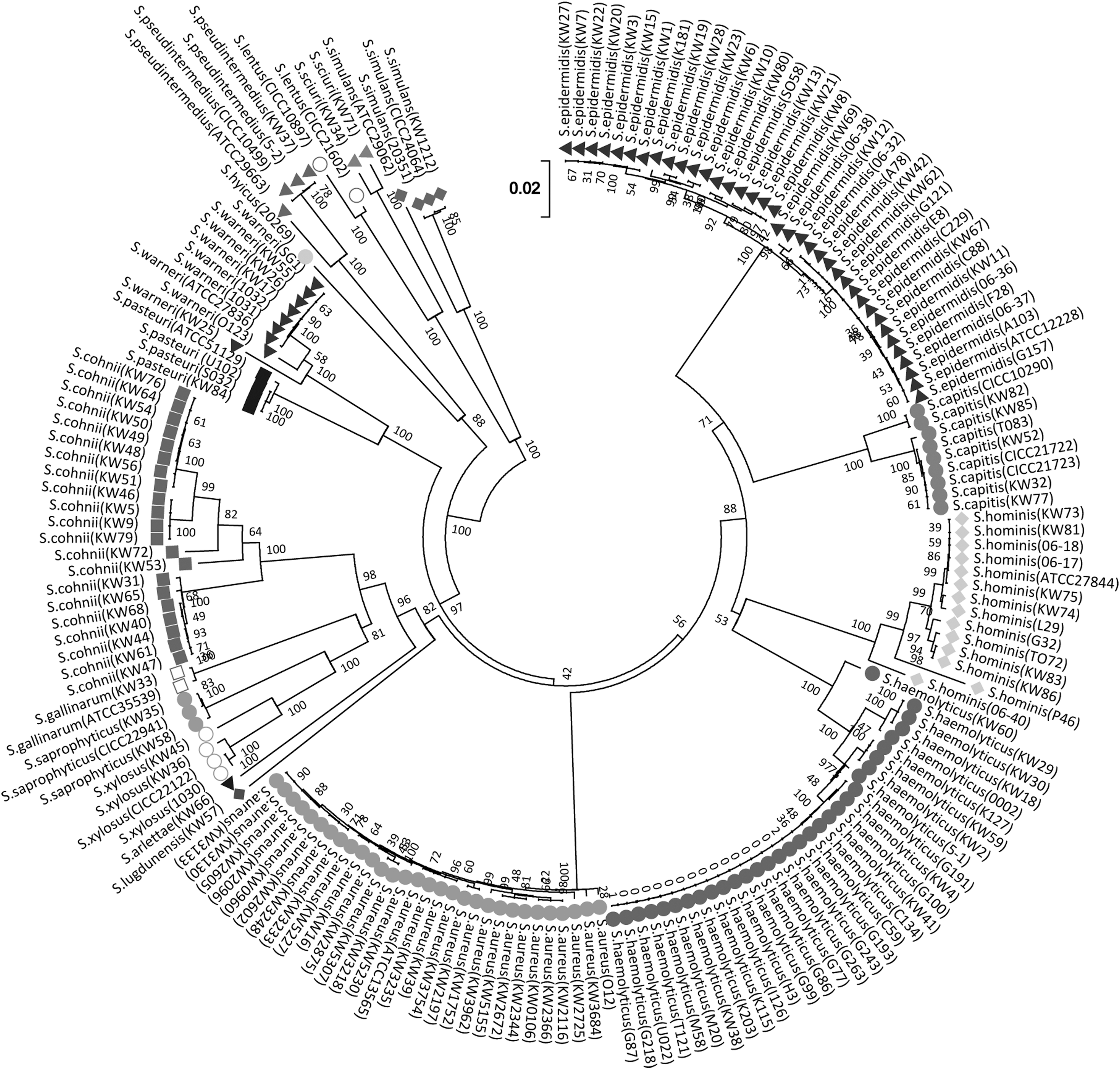
Neighbor-joining tree illustrating the phylogenetic relationships of 180 Staphylococcus isolates based on the sequences of 6 housekeeping genes (femA, ftsZ, gap, pyrH, rpoB, and tuf). Evolutionary analysis was conducted using MEGA7 and sequence distances were computed using the maximum composite likelihood method. The default cutoff value for neighbor-joining tree was 50%. All the positions containing gaps or missing data were eliminated, which resulted in a total of 3054 positions in the final dataset. The strains belonging to the same species were labeled with identical color and symbol. ATCC, American Type Culture Collection; CICC, China Center of Industrial Culture Collection.
Comparative Genotyping Result of 180 Staphylococcus Strains by Comprehensive Multilocus Sequence Typing (MLST) and MLST Methods
The loci of arcC, gtr, and tpiA were not amplified from 3 S. epidermidis strains by the primers used in the specific-dependent MLST scheme; hence the remaining 34 S. epidermidis strains were typed into 30 sequence types.
MLST, multilocus sequence typing.
Comparison of the comprehensive MLST and MLST of Staphylococcus strains
The discriminatory power of the comprehensive MLST scheme was compared to the species-dependent MLST schemes available for S. aureus, S. epidermidis, S. haemolyticus, and S. hominis (Table 4). By the species-dependent MLST, 30 S. aureus strains were typed into 17 sequence profiles, 32 S. haemolyticus strains were typed into 12 sequence profiles, and 14 S. hominis strains were typed into 12 sequence profiles. For S. epidermidis, 34 strains were typed into 30 sequence profiles, but 3 strains failed in PCR amplification of the arcC, gtr, or tpiA loci. Although the species-dependent MLST had slightly higher discriminatory power than the comprehensive MLST, the newly developed scheme could be universally applied to the identification and genotyping of various species of Staphylococcus strains.
Discussion
Due to increasing clinical significance of CoNS strains, rapid and accurate identification of Staphylococcus at species level are essential for epidemiological surveillance and precise determination of host-pathogen relationships (Becker et al., 2014). Culture-based identification of CoNS strains consisting of diverse species appears to be unreliable due to interspecies phenotype similarity and limited information in databases (Hwang et al., 2011). Consistent with previous reports (Hwang et al., 2011), our results showed that closely related S. pasteuri and S. warneri species were not differentiated by the phenotypic identification system of VITEK 2.
With the rapid advances in sequencing techniques and bioinformatics data analysis, genotyping identification of CoNS has emerged as a superior tool for clinical diagnosis (Zadoks and Watts, 2009). Supported with a large amount of sequence data in public databases and guidelines for data interpretation, 16S rRNA is the most commonly used target for bacterial identification and phylogenic study (Hwang et al., 2011). However, previous studies showed that 16S rRNA sequence analysis provided poor identification of certain CoNS species, such as S. caprae and S. capitis (Shah et al., 2007; Hwang et al., 2011), which was consistently supported by our results. Housekeeping genes possess higher nucleotide variations and could provide better discriminatory power than 16S rRNA sequence for genotyping CoNS species. Sequence variations collected from multiple independent genes could more truly reflect the genetic relatedness of the isolates than single target data (Perez-Losada et al., 2013). In this study, we selected and evaluated alternative genes rpoB (Drancourt and Raoult, 2002), tuf (Hwang et al., 2011), femA (Vannuffel et al., 1999), and gap (Yugueros et al., 2001), along with housekeeping genes ftsZ (encoding cell division protein) and pyrH (encoding uridylate kinase) for the species-level identification of Staphylococcus strains. Sequence diversity analysis of 180 Staphylococcus strains of 18 species showed that the alternative targets had significantly higher numbers of alleles, polymorphic sites, and nucleotide diversity (π) than 16S rRNA.
As a rapidly developing technology, whole genome sequencing has become the most powerful method for the identification and epidemiological investigation of microbial pathogens. Currently, the cost is still high and data analysis remains challenging for routine clinical diagnosis of Staphylococcus strains (Ronholm et al., 2016). MLST is a high-resolution genotyping method for identifying species and genus of bacteria, studying the genetic relatedness among strains, and evaluating the association between genotype and disease (Didelot and Maiden, 2010). To overcome the limitation of currently published MLST schemes only applicable to a few species of Staphylococci, we herein designed and evaluated a new MLST scheme, which was universally applied to 18 different staphylococcal species. By sequencing the internal DNA fragments of the six housekeeping genes with allelic polymorphisms, we achieved sufficient discriminatory power for the identification and genotyping of 180 Staphylococcus isolates.
Furthermore, the combined multitarget sequence was used in the study of genetic diversity of Staphylococcus strains belonging to different species. Comprehensive MLST of 180 Staphylococcus isolates generated 103 different sequence profiles, indicating great genetic diversity of the strains. Compared with previously developed species-dependent MLST schemes for S. aureus, S. epidermidis, S. haemolyticus, and S. hominis, the comprehensive MLST scheme had similar levels of discriminatory power. In addition, this new scheme provided good discrimination for typing some opportunistic species, including S. cohnii, S. capitis, and S. warneri, which overcome the lack of efficient genotyping methods for CoNS species. Nevertheless, the comprehensive MLST displayed variable discriminatory power for different species of Staphylococcus, for example, high discriminatory power (D = 0.955) for 37 S. epidermidis strains, but relatively low discriminatory power (D = 0.716) for 32 S. haemolyticus strains. This could be due to the inconsistent sequence variation rates of the target genes and unbalanced number of strains in each species, which needs to be confirmed by further testing a larger number of diverse strains.
Conclusions
In this study, a comprehensive MLST scheme utilizing six housekeeping genes was developed for species identification and genotyping of Staphylococci. Sequence diversity analysis of the target genes produced reliable results for the identification and genotyping of 180 Staphylococcus strains belonging to 18 species. Phylogenetic analysis of the combined target sequences led to a better understanding of the evolutionary relationship of Staphylococcus strains, especially the pathogenic CoNS.
Footnotes
Acknowledgments
This work was supported by the grants from Ministry of Science and Technology of China (National Key R&D Program China, 2018YFC1603900), Science and Technology Commission of Shanghai Municipality (16DZ0500202), and Chinese Pharmacopoeia Commission (standard improvement project of 2015 and 2016). We thank Mrs. Sue Reed in the Molecular Characterization of Foodborne Pathogens Research Unit at the USDA, ARS, ERRC, for reviewing the article.
Disclosure Statement
There are no conflicts of interest to declare.