
Abstract
Listeria monocytogenes is an opportunistic human foodborne pathogen that causes severe infections with high hospitalization and fatality rates. Clonal complex 9 (CC9) contains a large number of sequence types (STs) and is one of the predominant clones distributed worldwide. However, genetic characteristics of ST477 isolates, which also belong to CC9, have never been examined, and little is known about the detail genomic traits of this food-associated clone. In this study, we sequenced and constructed the whole-genome sequence of an ST477 isolate from a frozen food sample in China and compared it with 58 previously sequenced genomes of 25 human-associated, 5 animal, and 27 food isolates consisting of 6 CC9 and 52 other clones. Phylogenetic analysis revealed that the ST477 clustered with three Canadian ST9 isolates. All phylogeny revealed that CC9 isolates involved in this study consistently possessed the invasion-related gene vip. Mobile genetic elements (MGEs), resistance genes, and clustered regularly interspaced short palindromic repeats (CRISPR)/Cas system were elucidated among CC9 isolates. Our ST477 isolate contained a Tn554-like transposon, carrying five arsenical-resistance genes (arsA–arsD, arsR), which was exclusively identified in the CC9 background. Compared with the ST477 genome, three Canadian ST9 isolates shared nonsynonymous nucleotide substitutions in the condensin complex gene smc and cell surface protein genes ftsA and essC. Our findings preliminarily indicate that the extraordinary success of CC9 clone in colonization of different geographical regions is likely due to conserved features harboring MGEs, functional virulence and resistance genes. ST477 and three ST9 genomes are closely related and the distinct differences between them consist primarily of changes in genes involved in multiplication and invasion, which may contribute to the prevalence of ST9 isolates in food and food processing environment.
Introduction
L
According to the Food and Drug Administration, over the past 2 years, extensive food products have been recalled due to the suspicions of L. monocytogenes contamination. Recent studies indicated multinational listeriosis outbreak due to L. monocytogenes transmission (Jackson et al., 2016; Angelo et al., 2017). Another study isolated L. monocytogenes in ∼20% of 1036 raw food samples from 24 Chinese cities from South to North China (Wu et al., 2015). In China, the predominant sequence types (STs) isolated from food are ST9, ST8, and ST87 (Wang et al., 2012; Chen et al., 2018). ST9 isolates, which belong to clonal complex 9 (CC9), were also found to infect immunodeficient humans and cause bacteremia (Ragon et al., 2008). However, potential pathogenicity of CC9 strains has not been performed previously.
L. monocytogenes chromosomal genes are highly conserved with respect to their structural organization (Kuenne et al., 2013). The diversity and wide variation are mainly reflected in their accessory genomes, although they only occupy a small fraction (12–23%) of the L. monocytogenes gene content compared with the core genomes (den Bakker et al., 2013). Accessory genetic materials, including mobile genetic elements (MGEs) and the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas system, contribute to the phenotypic plasticity and adaptability of this pathogen (Kuenne et al., 2013). The Tn554-like transposon that introduces an arsenate resistance operon (arsCBADR) into the L. monocytogenes chromosome has been reported (Kuenne et al., 2013) but has not been characterized in detail. Studies on the structural organization of the Tn554-like transposon and genetic information are therefore necessary to assess the transmission and pathogenicity among L. monocytogenes isolates.
In the present study, we sequenced and analyzed the genetic sequence of an ST477 isolate and compared it with previously sequenced genomes of L. monocytogenes from human, animal, and food sources to (1) identify the genetic characteristics of the ST477 isolate; (2) investigate the genetic diversity of CC9 isolates from different sources and distant geographical regions; and (3) examine genome-level variation among CC9 isolates. Our results would serve as a framework for future analyses of CC9 strains.
Materials and Methods
Isolate
The ST477 L. monocytogenes isolate (NH1) with the 1/2c serotype examined in this study was originally isolated from a quick-frozen food sample consisting of wheat flour, and meat was purchased in Hebei province, China, in 2010. The isolate displayed resistance to multiple antibiotics, including tetracycline, erythromycin, chloramphenicol, streptomycin, cefotaxime, and trimethoprim/sulfamethoxazole (Yan et al., 2010), as well as cotolerance to cadmium and benzalkonium chloride (Xu et al., 2014).
Genome sequencing and annotation
Genomic DNA was extracted from overnight culture grown in a brain/heart infusion broth at 37°C using the DNeasy DNA extraction kit (Axygen, Union City). Purified genomic DNA was sequenced using the PacBio RS II (Pacific Biosciences, Menlo Park, CA) and Illumina MiSeq (Illumina, San Diego, CA) platforms as previously described (Zheng et al., 2017). A single contiguous chromosome consisting of 3,002,491 bp and a 37.92% GC, DNA G+C content was obtained (Supplementary Fig. S1). The complete sequence of NH1 was deposited in GenBank under the accession number CP021325. Gene prediction and annotation were performed as previously described (Zheng et al., 2017).
Phylogenetic and clustering analyses
We constructed a phylogenetic tree of the ST477 isolate and 58 previously published genome sequences with different geographic areas and sources to investigate the genomic traits of the ST477 isolate (Supplementary Table S1). These 58 isolates include 25 human-associated, 27 food-associated, and 6 from other or unknown sources isolates, and comprised 6 CC9 and 52 other clones. This data set comprised two genomes from lineage III to root the phylogenetic tree. The tree was constructed under a maximum likelihood optimality criterion using RAxML v7.4.2 (Uhlemann et al., 2017).
In silico MLST analysis and determination of profiles of virulence and resistance genes
Clonal complexes were determined by the Institut Pasteur MLST database with default parameters (
Analysis of the features of CC9 genomes harboring Tn554-like transposon
Based on the results of BLASTN search, eight isolates that harbored Tn554-like transposon and two isolates that did not were analyzed to compare the similarities of their genomic contents. Antimicrobial and heavy metal resistance genes, stress survival islet 1 (SSI-1), and IS3 were examined using a BLASTX search as described previously (Soge et al., 2016). PHASTER (
Whole-genome alignment
Whole-genome alignment of seven CC9 genomes was performed. Mauve software was used to align the genomic sequences and identify locally collinear blocks shared by all genomes (Ortiz et al., 2016).
Single-nucleotide polymorphism identification and analysis
The WGS fastq data of six CC9 isolates were aligned against the chromosome of the ST477 to detect single-nucleotide polymorphisms (SNPs) using GATK and SAMtools as previously described (Cheng et al., 2014).
Results
Phylogenetic analysis of ST477 isolate
We determined a core gene sequence-based phylogeny based on 2037 single-copy orthologs present in the 59 genome sequences (Fig. 1). This tree resolved three lineages (lineage I–III). Unsurprisingly, the two lineage III isolates chosen as outgroup were clearly distant from all other isolates, whereas lineages I and II were strongly structured into major subgroups, each comprising multiple closely related isolates. Phylogenetic lineages exhibited strong association with serotype (Fig. 1). Lineage I contained two major clades: one consisted predominantly of serotype 4b plus one sample of serotype 4e and one of unknown serotype; in the other clade, the majority (6/11) of samples were of unknown serotype, the other samples belonged to serotypes 1/2b, 7, and one sample of serotype 1/2a. Lineage II consisted predominantly of serotype 1/2a, but contained two clades composed of serotypes 1/2c and 3c and a clade that included one serotype 3a isolate and an isolate of unknown serotype. The subgroups did not show any clear association with isolate source, although uncertainty of the infection source for the human clinical isolates could obscure such patterns. Also, isolates from one country did not cluster together phylogenetically.
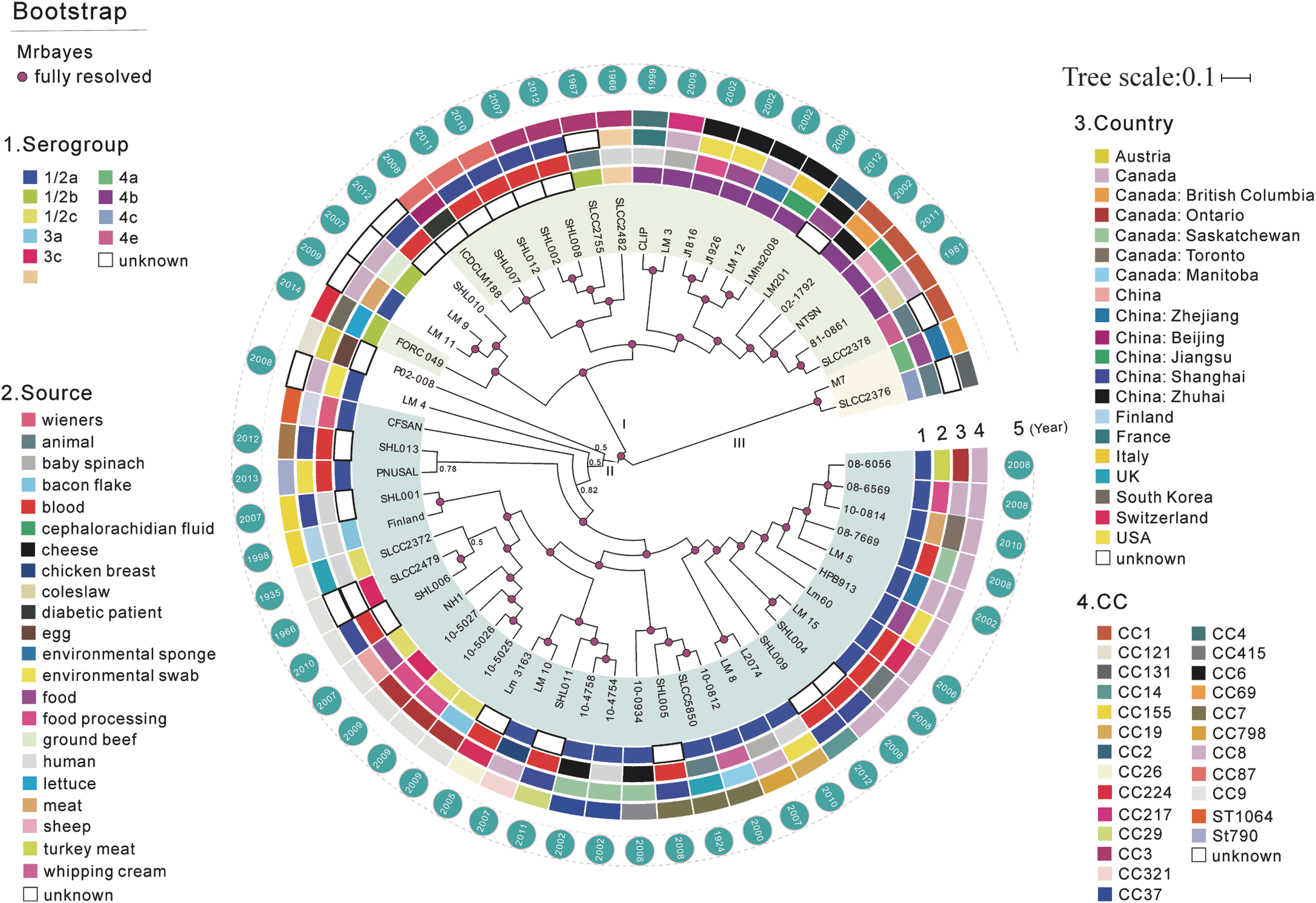
Maximum-likelihood phylogeny of 59 Listeria monocytogenes isolates based on 2073 single-copy core genes. The annotation rings surrounding the tree, from inside to outside, depict (1) serotype, (2) sample isolation source, (3) geographic origin, (4) clonal cluster number, and (5) year of sample collection. Shading behind the branches represents lineage, and white indicates unknown information.
Although most of the 59 isolates were distributed among 35 known MLST types (Supplementary Table S1), we discovered that four displaying novel STs beared undescribed MLST alleles or previously unreported allelic combinations: three of these isolates, including two Canadian isolates from meat products and one clinical isolate from China, formed a clade within lineage I; the other isolate (an environmental sample from Canada) occupied a basal position in lineage II. Seven CC9 isolates derived from food and clinical sources formed a single clade within lineage II. In the CC9 subgroup, the ST477 isolate displayed the closest relationship to three ST9 food-associated isolates from Canada (10-5025, 10-5026, and 10-5027).
Biological features of different L. monocytogenes clonal complexes
Certain variations in virulence and stress resistance genes among L. monocytogenes lineages and subgroups were observed (Fig. 2). As expected, the major pathogenicity island LIPI-1 was highly conserved. Complete LIPI-3 and the LIPI-4 islands were almost exclusively detected within lineage I. The number of internalins ranged from two (CFSAN004330) to nine in the examined genomes. InlG was absent in lineage I except the CC6 subgroup. The ST477 isolate contained LIPI-1, nine inl genes, but not the LIPI-3 and LIPI-4 islands. Overall, there was rare difference between the ST477 and other CC9 isolates, except that the ST477 isolate lacked the bile-resistance gene mdrM. In addition, similar to clinical isolates, all CC9 isolates harbored inlH and inlK. Strikingly, a virulence gene (vip), encoding a surface protein, was universally present in CC9 isolates. Collectively, these results demonstrate the heterogeneity of biological features among L. monocytogenes subgroups and mark frequent virulence genes in CC9 clone.

Virulence and resistance profiles across the phylogeny of the 59 L. monocytogenes isolates.
Genome comparisons of CC9 isolates
We identified a Tn554-like transposon sequence in the ST477 isolate. This transposon encodes three consecutive transposase genes (tnpABC), five arsenate resistance genes, and four genes encoding hypothetical proteins with unknown function (Fig. 3). Through BLASTN searches in GenBank using this nucleotide sequence as the query, highly conserved homologues (99% nucleotide sequence similarity) in eight other L. monocytogenes and two Listeria ivanovii genomes were found. The Tn554 transposon from Staphylococcus aureus and the Tn554-like transposon from the ST477 isolate shared a modular composition with a transposase module (consisting of the transposases TnpABC) and different resistance markers. Transposase genes of the Tn554-like transposon shared a low degree of similarity with the Tn554 transposon. Unlike the Tn554-like transposon, the Tn554 transposon is chromosomally integrated within the radC gene and displays a 5′-GATGTA-3′ and 5′-CAAGTT-3′ sequence at its left- and right-end junction, respectively. Thus, there are important differences between these two transposons. The GC content of Tn554 (32.6%) and Tn554-like (37.9%) transposons was similar to that of S. aureus and L. monocytogenes genomes (32.6–33% and 37.8–38%, respectively), suggesting that the ancestor of the Tn554-like transposon probably arose from L. monocytogenes rather than S. aureus. Not all CC9 isolates carry the Tn554-like transposon; however, eight isolates containing this transposon belong to CC9 clone, suggesting a single event of insertion into the CC9 background.
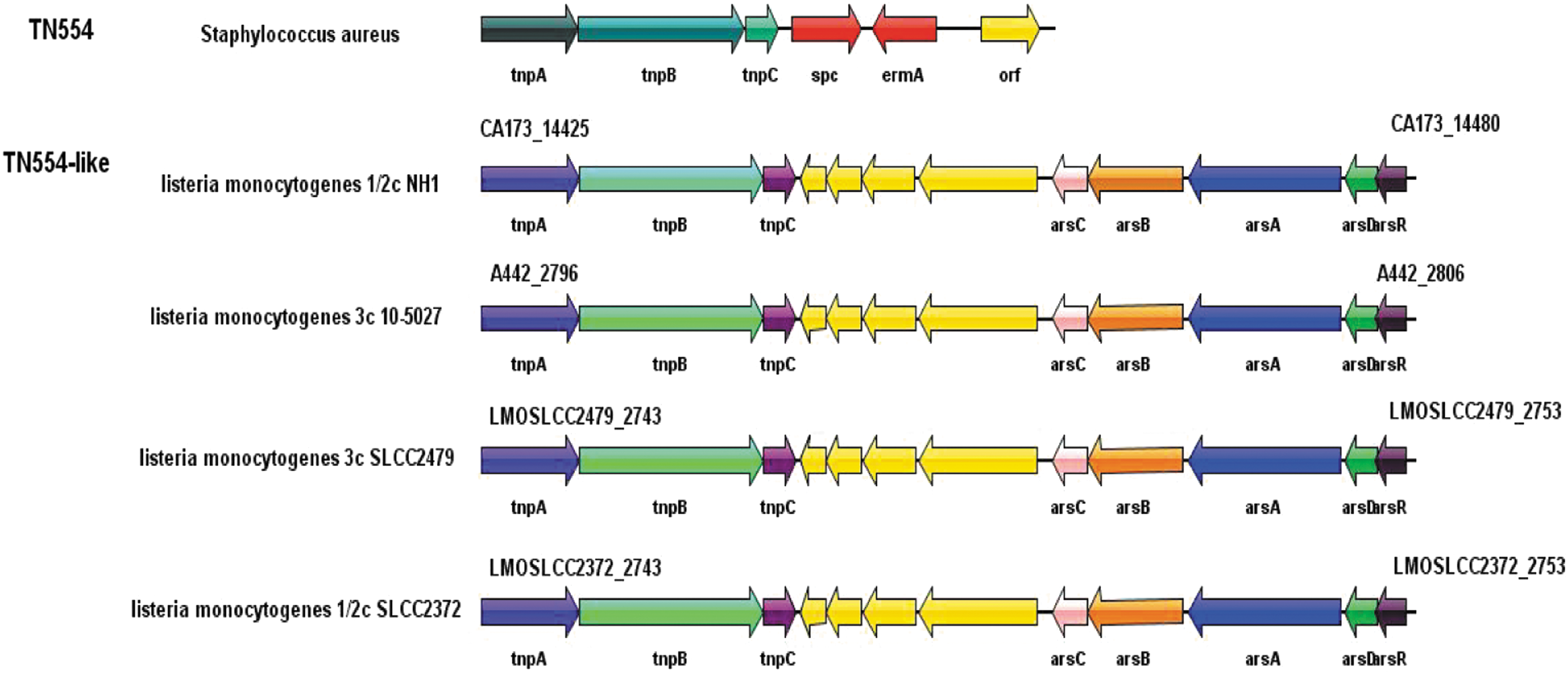
Organization of the L. monocytogenes Tn554-like transposon and comparison with the related transposon Tn554. ORFs are shown as arrows, indicating the transcription direction, and the colors of the arrows represent different fragments. Proteins with other or unknown functions are shown in yellow.
SSI-1 had been postulated to facilitate elevated resistance to several types of stress in certain L. monocytogenes strains. Our results indicated that no specific patterns existed among these strains (Table 1); however, both the ST477 isolate and two Canadian isolates (10-5026 and 10-5027) harbored this islet, further emphasizing their similarity.
Relevant Features of the Listeria monocytogenes Strain Genomes
The presence of mobile genetic elements is indicated by the “+” sign and the absence is indicated by the “−” sign.
GC, DNA G+C; N.A., unknown information; ST, sequence type.
PHASTER-based analysis of the fully closed ST477 genome predicted four putative complete prophages (Supplementary Table S2), including three common prophages (A006, B025, and LP_101) and a composite Φlmc1 prophage (A118, A006, and B025). Highly similar (99–100% identity) Φlmc1 sequences, including the site-specific integrase region, were detected in only two L. monocytogenes isolates (SLCC2482 and AT3E), isolated from humans and food from the United States and Finland, respectively (Table 1). Although the three Canadian isolates were devoid of this prophage, they still harbored the site-specific integrase region (Fig. 4), suggesting a possible recombination event that inserted the prophage into their chromosomes.

Linear comparison of the Φlmc1 prophage in four different L. monocytogenes isolates. The prophage is present in NH1, SLCC2482, and AT3E strains, and is absent from strain 10-5027. Homologous gene clusters in different isolates are shaded in gray (>97% nucleotide sequence similarity).
Our ST477 genome harbored resistance genes against several types of antibiotics, including sulfonamides (folP, thyA), aminoglycosides (adeC, strA), glycopeptides (cls), lincosamides (lmrB), and fosfomycin (fosX) (Table 2). In addition, this genome contained three efflux pump-related genes that confer resistance to tetracyclines (tetA) and quinolone (fepA and norB). Moreover, the small multidrug resistance (SMR) transporter qacE was identified in the ST477 and other CC9 strains, increasing their capacity to colonize food production plants. Notably, the antibiotic and heavy metal resistance genes were identical to those found in CC9 isolates, but greatly differed from those of non-CC9 isolates. The presence of multiple resistance genes in CC9 genomes further reiterates the health risk as these strains could be transmitted to humans via food contamination.
Comparison of Resistance Genes in Listeria monocytogenes from Different Countries
The presence of resistance genes is indicated by the “+” sign and the absence is indicated by the “−” sign.
MFS, major facilitator superfamily; MATE, mulitidrug and toxic compound extrusion; SMR, small multidrug resistance.
The ST477 and three closely related isolates contained an identical CRISPR cluster, consisting of four spacer sequences interspersed between five highly conserved 29 bp direct repeat regions, and were not associated with any cas genes (Supplementary Table S3). All CC9 isolates shared identical spacer arrays, which were directed versus known listeriaphages (B054, P70). However, the number of spacers in CC9 was much less than other clones.
SNP analysis among CC9 isolates
The initial comparative analysis of the ST477 genome with three closely related ST9 sequences was performed following the genome alignment of seven CC9 isolates using Mauve (Supplementary Fig. S2). The NH1 genome is ∼37 kbp larger than the 10-5027 genome and 35 kbp larger than the other two genomes (Supplementary Table S4). Annotation of the predicted coding sequence regions for the four genomes using the NCBI database revealed that the ST477 genome contained the highest number of genes (n = 3084) and pseudogenes (n = 24). The ST477 genome was highly similar to the three ST9 isolates, except for two major regions, ranging from ∼669–705 and 2754–2761 kbp in ST477 (Fig. 5). These regions contain genes that encode hypothetical proteins, metabolism enzymes, and phage-related proteins. Thus, the ST477 isolate and these Canadian ST9 isolates may share a common gene pool.
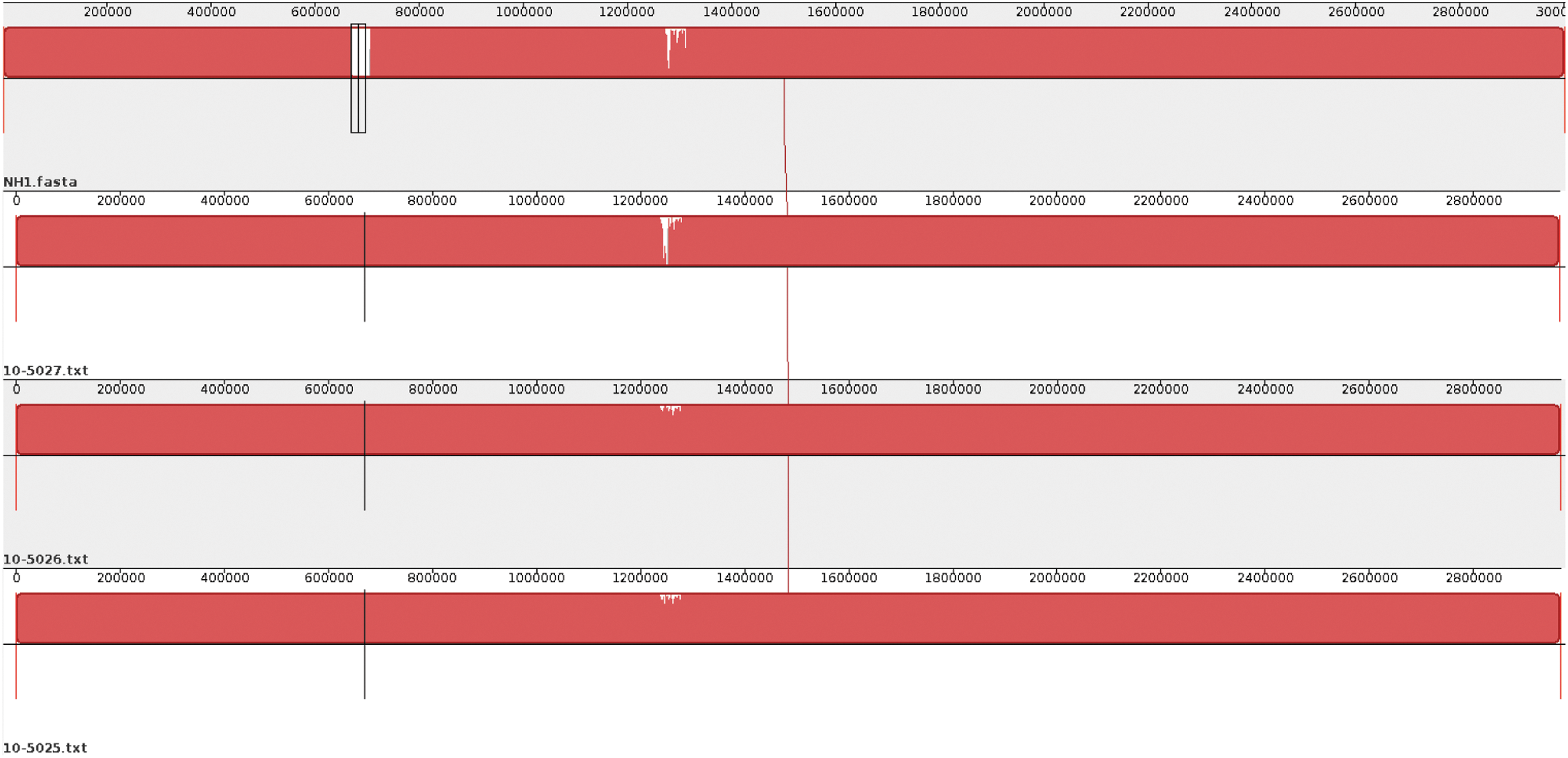
Whole-genome alignment of L. monocytogenes using the Mauve, version 2.3.1, and the figure was generated using the Mauve viewer. Larger white portions identify areas of low similarity. Areas that are completely white within a block are not aligned and most likely contain sequence elements specific to a particular genome.
Variant detection, based on read mapping of each of the CC9 isolates to our ST477 isolate, showed that 291 nonsynonymous SNPs occurred in ST477 genome (Supplementary Table S5). A further comparison of SNPs among three closely related Canadian ST9 isolates indicated that 60 nonsynonymous substitutions were shared by these isolates (Table 3). Of note, nonsynonymous substitutions occurred in important functional genes smc, ftsA, and essC and other genes encoding cell surface proteins. We also identified nine different indels in each of the Canadian isolates. However, only one indel encoding a hypothetical protein was identified in all of these isolates. This result showed that these genes play key role in the evolution pattern of ST477 and ST9.
Single-Nucleotide Polymorphisms and Indels Shared in Three Canadian ST9 Isolates
Refers to genomic nucleotide positions and gene products from the mapping reference sequence of the novel ST477 isolate.
MBL, metallo β-lactamase; MFS, major facilitator superfamily; SMC, structural maintenance of chromosomes; SNP, single-nucleotide polymorphism.
Discussion
CC9 isolates are frequently isolated in various kinds of foods (Yan et al., 2010; Wu et al., 2016). This is one of the clones with the highest geographical transition rates (Chenal-Francisque et al., 2011). However, its genetic characteristics and pathogenesis are poorly understood. Whole-genome sequence of ST477 isolate has never been reported as of now. The phylogenetic analysis shows that strains from one country did not cluster together phylogenetically, which is consistent with the previous observation that L. monocytogenes clones could have broad geographic distributions (Chenal-Francisque et al., 2011). Furthermore, ST477 isolate has a close evolutionary relationship with three other ST9 isolates from Canada.
The virulence potential of clones is determined, at least in part, by vertical transmission (Maury et al., 2016). Thus, the distribution and variation of known virulence determinants can not only suggest important differences among strains regarding virulence potential but also indicate the evolutionary patterns among L. monocytogenes lineages and subgroups. Notably, all CC9 strains consistently carried vip and nine inl genes, whereas these genes were scattered among non-CC9 strains. The Vip protein is required for entry into some mammalian cells (Martins et al., 2012). Internalins play essential roles in host cell invasion and virulent phenotypes in animals (Balandyte et al., 2011). Of which, internalin G and H are more important for the survival of L. monocytogenes. Gene inlK was previously described to play an important role in escape of autophagic recognition (Dortet et al., 2012). Together, these results provide preliminary evidence that CC9 isolates have at least a theoretical pathogenic potential and could pose a significant threat to the public health.
Notably, we identified the exclusive presence of Tn554-like transposon in the CC9 isolates, which has been reported in a previous study (Kuenne et al., 2013) but has not been characterized in detail. To our knowledge, only a few Tn554-related transposons have been identified to date: Tn6188 is the only transposon identified in L. monocytogenes that confers tolerance to disinfectants. Tn558, Tn559, and Tn5406, found in various Firmicutes isolates, confer resistance to antibiotics (Müller et al., 2013). Tn554 and related transposons are neither flanked by direct or inverted repeats, nor generate a duplication of the target sequence at their integration site (Murphy and Löfdahl, 1984). Whole-genome sequencing demonstrated that the Tn554-like transposon contains five arsenic-resistance genes, which are present in the resistant phenotype. However, subsequent functional studies of this transposon are necessary. In addition, the implication from this study, in which the emergence of this transposon may be linked to the CC9 background, deserves further investigation.
SSI-1 is a five-gene cluster known to enhance the adaptation of L. monocytogenes to food environments based on deletion mutant studies (Ryan et al., 2010). However, no significant differences in the stress tolerances of naturally occurring L. monocytogenes isolates with or without SSI-1 were found (Hingston et al., 2017). The former mechanism appears less probable given that CC9 isolates are prevalent in foods but lack the SSI-1 cluster.
The distribution of different spacers indicates that the seven CC9 strains compared in this work were infected by the listeriaphages B054 and P70. Self-defense relies on the Cas enzymatic machinery (Barrangou et al., 2007), the absence of cas genes would likely render the entire CRISPR system nonfunctional. Thus, CC9 isolates appear more likely to acquire foreign genetic elements, including antibiotic resistance genes, and pose a greater risk for public health compared with other isolates containing the cas genes.
Whole-genome comparison indicated that the ST477 isolates and the three Canadian isolates show high genomic similarity and may share a common gene pool. Further inspection of SNPs showed that most of nonsynonymous mutations presented in metabolism genes. A previous study showed that smc played a central role in chromosome condensation and segregation to affect the bacterial growth (Wang et al., 2014). Another study indicated that ftsA encoded cell division protein essential for the survival of the bacteria (Furusato et al., 2018). Moreover, essC and other genes encoding cell surface proteins played important roles in the capacity of invasion to the host cell (Martins et al., 2012; Pinheiro et al., 2017). The common ancestor of ST477 and ST9, during the course of its evolution, has continuously evolved their metabolism and physical characteristics to adapt to different environments. This may also explain why ST9 isolates are more prevalent in food products.
Conclusions
The present study preliminarily suggests that the wide distribution of CC9 clone in various kinds of foods is likely due to the presence of MGEs and functional virulence and resistance genes. Our ST477 isolate has close evolutionary relationship with ST9 isolates. Genomic differences between ST477 and ST9 include changes in the genes for origin segregation and cell surface protein, suggesting these genes may contribute to the adaption/colonization of L. monocytogenes in food processing environments. Further research should be carried out to confirm the evolutionary relationships between ST477 isolates and pathogenicity of CC9 strains.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 31571934), the Science and Technology Planning Project of Guangdong Province, China (2014A020214001, 2016A020219001), the Fundamental Research Funds for the Central Universities, SCUT (D2170320), and the Central Universities construct the world first-class university (discipline) and Characteristic Development Guidance Special Fund (K5174960).
Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.