
Abstract
Gastroenteritis is a disease that can be caused by virulent strains of Vibrio parahaemolyticus in humans upon the consumption of contaminated seafood. In summer 2017, a sudden increase in the number of patients suffering from gastroenteritis due to a V. parahaemolyticus infection was observed at the Middle East Institute of Health University Hospital in Lebanon. The aim of this study was to analyze the isolates recovered from stool specimens, and to compare them using different phenotypic assays, genomic profiling techniques, and whole-genome sequencing, to achieve a better understanding of the current V. parahaemolyticus strains available in Lebanon. Virulence potential was analyzed based on the detection of the hemolysins: thermostable direct hemolysin (tdh), thermostable direct hemolysin-related hemolysin (trh), and thermolabile hemolysin (tlh). Resistance was determined by testing antibiotic susceptibility and performing PCR assays for β-lactamases and quinolone resistance determinants. Genetic relatedness was verified by multilocus sequence typing, pulsed-field gel electrophoresis, and whole genome-based single nucleotide polymorphism analysis. All of the isolates had the tdh +, trh −, group-specific PCR+ genotype, which is a characteristic of the O3:K6 pandemic clone. The isolates were resistant to ampicillin (100%), ceftazidime (86%), ticarcillin (14%), and amikacin (14%), belonged to the sequence type ST3, and had very similar phylogenetic fingerprints. The isolates undertaken in this study exhibited almost identical resistance, virulence, and phylogenetic patterns, confirming an outbreak linked to the spread of the pandemic O3:K6 serotype in the country.
Introduction
Vibrio parahaemolyticus is a Gram-negative, halophilic marine bacterium found in estuarine environments (Fujino et al., 1953). Most members of this species are nonpathogenic and can be found dominating the microbiota of the gut in fish (Hackney et al., 1980). However, virulent strains of V. parahaemolyticus can cause foodborne gastroenteritis in humans upon consumption of mishandled, undercooked or raw seafood (Fujino et al., 1953; Baker-Austin et al., 2008). This disease is usually self-limiting, and includes symptoms such as diarrhea, abdominal pain, vomiting, nausea, and low-grade fever (Lozano-Leon et al., 2003).
The thermostable direct hemolysin (tdh), thermostable direct hemolysin-related hemolysin (trh), and/or the thermolabile hemolysin (tlh) encode for toxins found in V. parahaemolyticus that are mostly responsible for the clinical symptoms seen in patients (Gonzalez-Escalona et al., 2017). Other virulence factors include two copies of the type III secretion system (T3SS), and two copies of the type VI secretion system (T6SS) (Makino et al., 2003; Yu et al., 2012).
In Lebanon, there are currently no reports about the incidence of V. parahaemolyticus infection. However, the presence of the species in seafood specimens in the Middle East has been detected at a rate of 1.76% and 3.8% in Lebanon and Egypt, respectively (Abdelnoor and Roumani, 1980; Edris et al., 2013).
In this study, we used whole-genome analysis to investigate a V. parahaemolyticus-linked outbreak, to determine the genomic profiles of the collected isolates, and to report the findings of the collected clinical V. parahaemolyticus isolates in Lebanon. The V. parahaemolyticus isolates were recovered from patients hospitalized in the Middle East Institute of Health that were suffering from gastroenteritis during summer 2017. Detailed molecular characterization was conducted on all recovered isolates using different approaches, including whole-genome sequencing (WGS) and revealed that the isolates belonged to ST3 O3:K6 serotype pandemic clone. This outbreak is particularly noteworthy in that it represents, to our knowledge, the first report of a virulent Vibrio complex emerging in Lebanon.
Materials and Methods
Ethics approval
Ethics approval was not required since the isolates were gathered and stored as part of routine clinical care. Patient records and information were anonymized before analysis.
Sample collection
All isolates were collected from the Middle East Institute of Health University Hospital in Lebanon over a period of 4 months, from July to October 2017. All the isolates were from patients suffering from gastroenteritis between the ages of 33 to 80, living in Matn, who recalled eating seafood. Isolates were designated as VP1-VP7 (Accession No.'s: PXIU00000000.1, PXIV00000000.1, PXIW00000000.1, PXIX00000000.1, PXIY00000000.1, PXIZ00000000.1, PXJA00000000.1, respectively). Isolates were first identified as Vibrio species based on the cellular morphology by the hospital staff, and all were subsequently included in the study.
DNA extraction
The clinical isolates were grown on tryptone soy agar plates with 3% NaCl and incubated overnight at 37°C. DNA was first extracted from the bacterial isolates by using the Nucleospin® Tissue Kit (by MACHEREY-NAGEL) according to the manufacturer's instructions.
Antibiotic susceptibility testing
The antibiotic susceptibility of the V. parahaemolyticus isolates was examined by the disk agar diffusion method in accordance with the guidelines of the Clinical and Laboratory Standards Institute (CLSI, 2010). Susceptibility testing was done using Muller-Hinton agar and a panel of 16 antibiotic disks, including cefepime, ceftazidime, ampicillin, piperacillin and tazobactam, ticarcillin, ciprofloxacin, amikacin, gentamicin, tobramycin, imipenem, meropenem, chloramphenicol, aztreonam, colistin, levofloxacin, and tetracycline.
Amplification of genetic markers, virulence, and resistance-encoding genes
Individual PCRs were performed for the genetic identifiers (toxR, pR72H, and orf8), and virulence genes (tdh, trh, and tlh) as listed in Supplementary Table S1. In addition, VPA0477 (a β-lactamase gene bla), VPA0095 (a gene encoding quinolone resistance qnr), aadA1 (aminoglycoside 3′-adenylyltransferase gene), strA (streptomycin resistance-encoding gene), and the β-lactamase genes, bla SHV, bla OXA, and bla TEM, were all amplified according to previous studies listed in Supplementary Table S1. PCRs were done using 10 × Taq buffer, 25 mM MgCl2, 5 U/μL of Taq polymerase, 2.5 mM deoxynucleoside triphosphate, 20 pmol/μL of each primer, 50 ng/μL of template DNA, and PCR gradient water. Products of the PCRs were visualized on a 1% agarose gel.
Pulsed-field gel electrophoresis
Pulsed-field gel electrophoresis (PFGE) was performed according to the PulseNet International protocol (
Whole-genome sequencing
Genomic libraries were constructed using the Nextera XT DNA Library Preparation Kit with dual indexing (Illumina, San Diego, CA). The kit was used to simultaneously fragment and tag the library, as per the manufacturer's instructions. The libraries were sequenced on an Illumina MiSeq with 250 bp × 2 read length.
Whole-genome assembly and annotation
Genome assembly was performed de novo using SPAdes Genome Assembler version 3.6.0 (Bankevich et al., 2012). Quality control was done using FastQC version 1.0.0 (Andrews, 2010). Illumina adapters and low-quality bases were clipped by TrimGalore (Krueger, 2015). The RAST server was used to annotate the resulting de novo assemblies (
The ResFinder 3.0 web server (
Core genome single nucleotide polymorphisms (cgSNPs) were called for each isolate using the Snippy pipeline (
BRIG 0.95 was used to create a circular comparative figure of the genomes with upper and lower identity thresholds of 100% and 95%, respectively (Alikhan et al., 2011). The whole-genomes of the collected isolates were aligned to that of RIMD 2210633, AQ3810, and AQ4037. RIMD 2210633 is a V. parahaemolyticus strain recovered from a patient with travelers' diarrhea at the Kansai International Airport quarantine station in 1996 (Makino et al., 2003). This strain is the current reference genome for the pandemic O3:K6 isolates. V. parahaemolyticus AQ3810 and AQ4037 are O3:K6 strains recovered from travelers at the Osaka Airport Quarantine Station in Japan in 1983 and 1985, respectively (Okuda et al., 1997).
Results
Antibiotic resistance, virulence, and typing
All the isolates were resistant to ampicillin, 86% to ceftazidime, and 14% for each ticarcillin and amikacin (Table 1). All were susceptible to cefepime, piperacillin and tazobactam, gentamicin, tobramycin, imipenem, meropenem, chloramphenicol, aztreonam, colistin, levofloxacin, and tetracycline. Moreover, all of the isolates were typed as ST3 according to the MLST 1.8 server (Larsen et al., 2012). This was additionally confirmed using the V. parahaemolyticus locus/sequence definitions database (University of Oxford, United Kingdom), with 100% alignment to each of the seven housekeeping genes.
Antibiotic Resistance Profile of the Vibrio parahaemolyticus Isolates
The antibiotic drugs shown in this figure are ceftazidime (CAZ), ampicillin (AMP), ticarcillin (TIC), ciprofloxacin (CIP), and amikacin (AMK).
I, intermediate resistance; R, resistance; S, sensitive; VP, Vibrio parahaemolyticus.
The detection of the species-specific toxR gene and pR72H sequence in all the isolates confirmed and verified the phenotypic identification. Moreover, the detection of the open reading frame orf8 in all the isolates revealed that all belonged to the pandemic strain O3:K6. In silico serotyping of the isolates was performed by alignment of the O3:K6 surface antigen EDM56499 to the sequenced whole-genomes. The fragment encoding the O antigen aligned with 618/621 (99%) of its corresponding loci in all isolates. Single nucleotide variants were identified at positions 291 (G>A), 345 (A>G), and 378 (G>A) in all of the isolates, showing no recombinational variation with complete dominance of the detected O3:K6 serovariant, and further confirming relatedness.
All the isolates were positive for tdh and tlh, and negative for trh, and accordingly had the following genotype: tdh +, trh −, tlh +. Only one resistance gene was detected in the isolates; gene corresponding to VPA0477, also known as a β-lactamase bla. VP1 showed intermediate resistance to ciprofloxacin (Table 1). VP1 contained a unique mutation at locus VP2987. The missense variant 331 A>T (Ser111Cys) codes for the cyaA gene encoding an adenylate cyclase. Previously it was shown that point mutation in cyaA resulted in kanamycin resistance through an altered adenylate cyclase and subsequently altered cyclic adenosine monophosphate (cAMP) receptor protein function (Mogre et al., 2017), and that reduced intracellular levels of cAMP arising from mutations in the cyaA gene downregulated the expression of the fosfomycin transporters (Karageorgopoulos et al., 2011).
The PFGE profiles of the isolates were determined, and the patterns obtained were analyzed. The generated dendrogram showed that all belonged to the same pulsotype and were grouped in three clusters except for VP1, which was untypable (Fig. 1). No mutations that could explain it being untypable were detected.
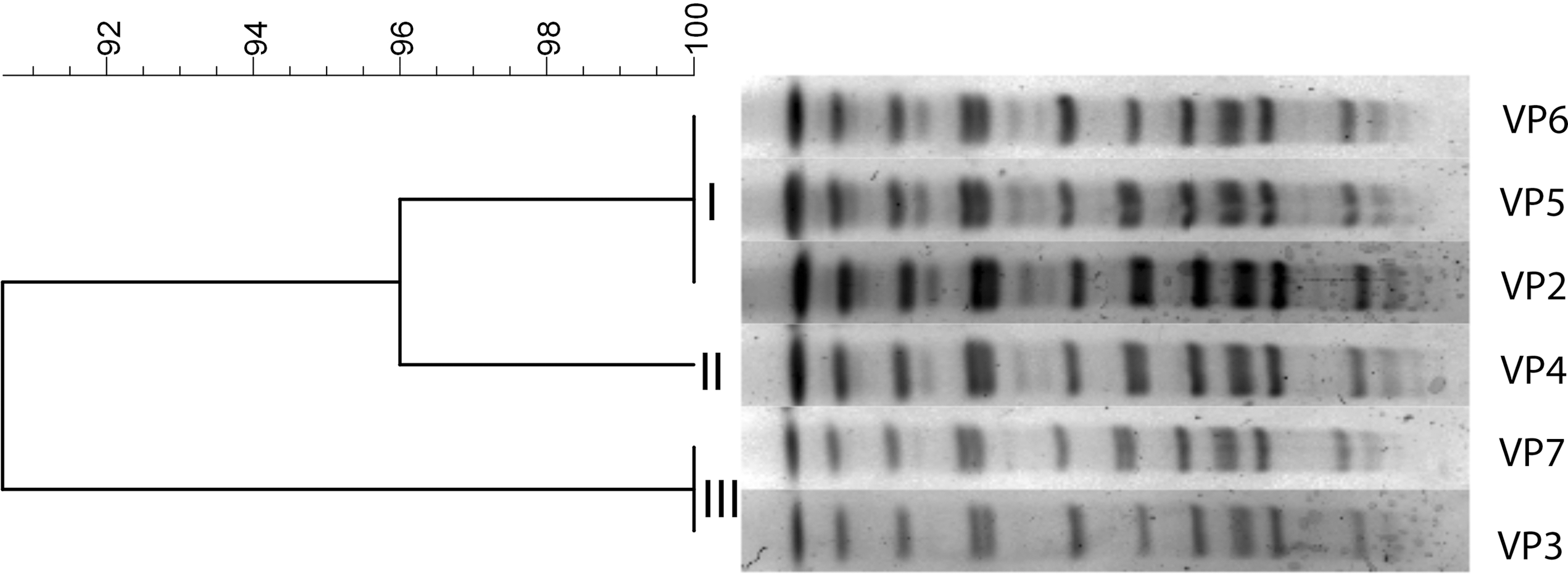
Dendogram showing the VP pulsotypes generated by PFGE using the restriction enzyme NotI. Image was generated using BioNumerics software (Applied Maths, Belgium). PFGE, pulsed-field gel electrophoresis; VP, Vibrio parahaemolyticus.
Pangenome analysis and SNP
The pangenome was generated using Roary and identified a total of 4870 genes. The core genome, which consists of genes shared by at least 99% of the isolates, included 4577 genes that were ubiquitous in all tested isolates. The accessory genome included 156 shell genes that were shared by 15–95% of isolates, and 137 cloud genes that were present in less than 15% of the isolates (Fig. 2). Additionally, two phylogenetic SNP-based trees were generated according to the core genome SNPs found in chromosome 1 (Fig. 3A) and chromosome 2 (Fig. 3B). The isolates had highly similar genetic makeup, except for few polymorphic loci. The phylogenetic trees for each chromosome were annotated at their branch points to highlight these distinguishing features. The three detected point mutations were synonymous, missense, and noncoding transcript variations (Table 2).
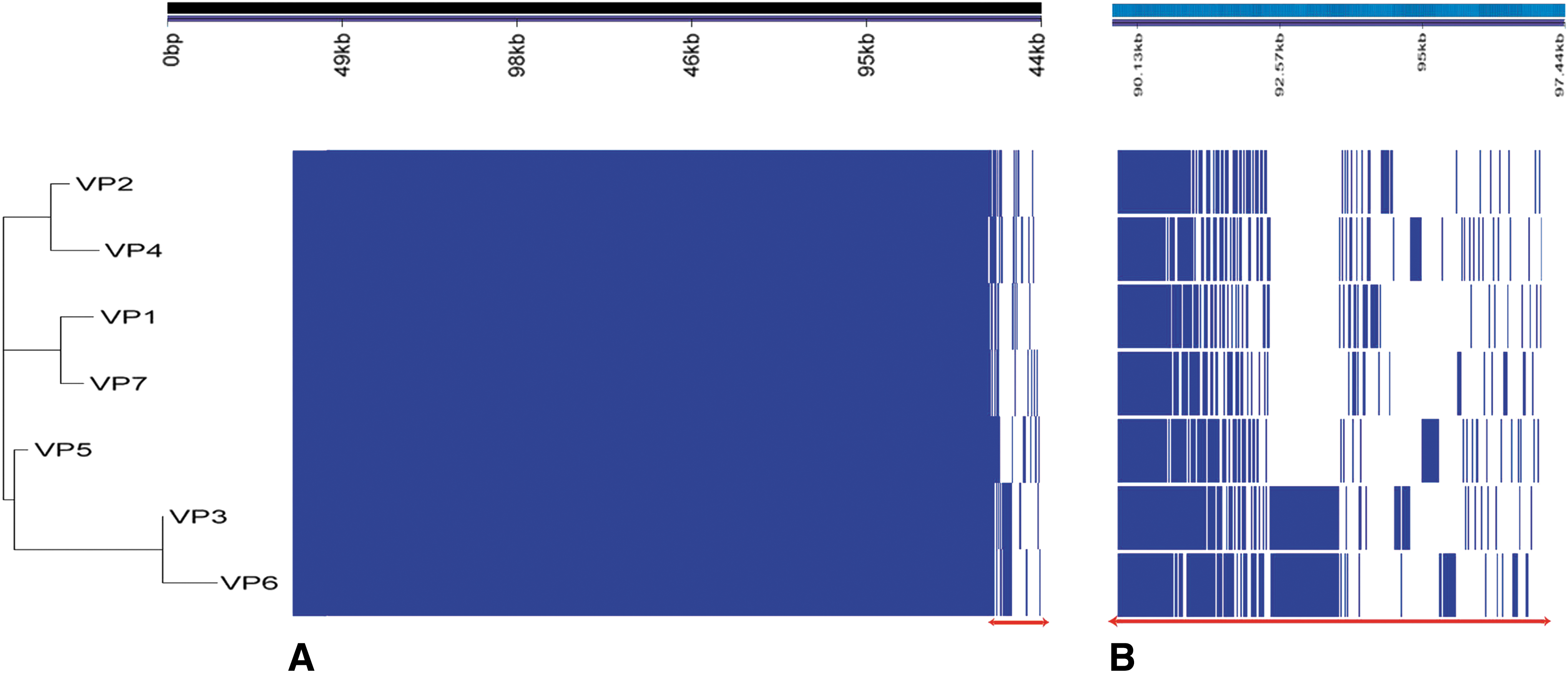
Pangenome analysis of the seven VP isolates using Roary
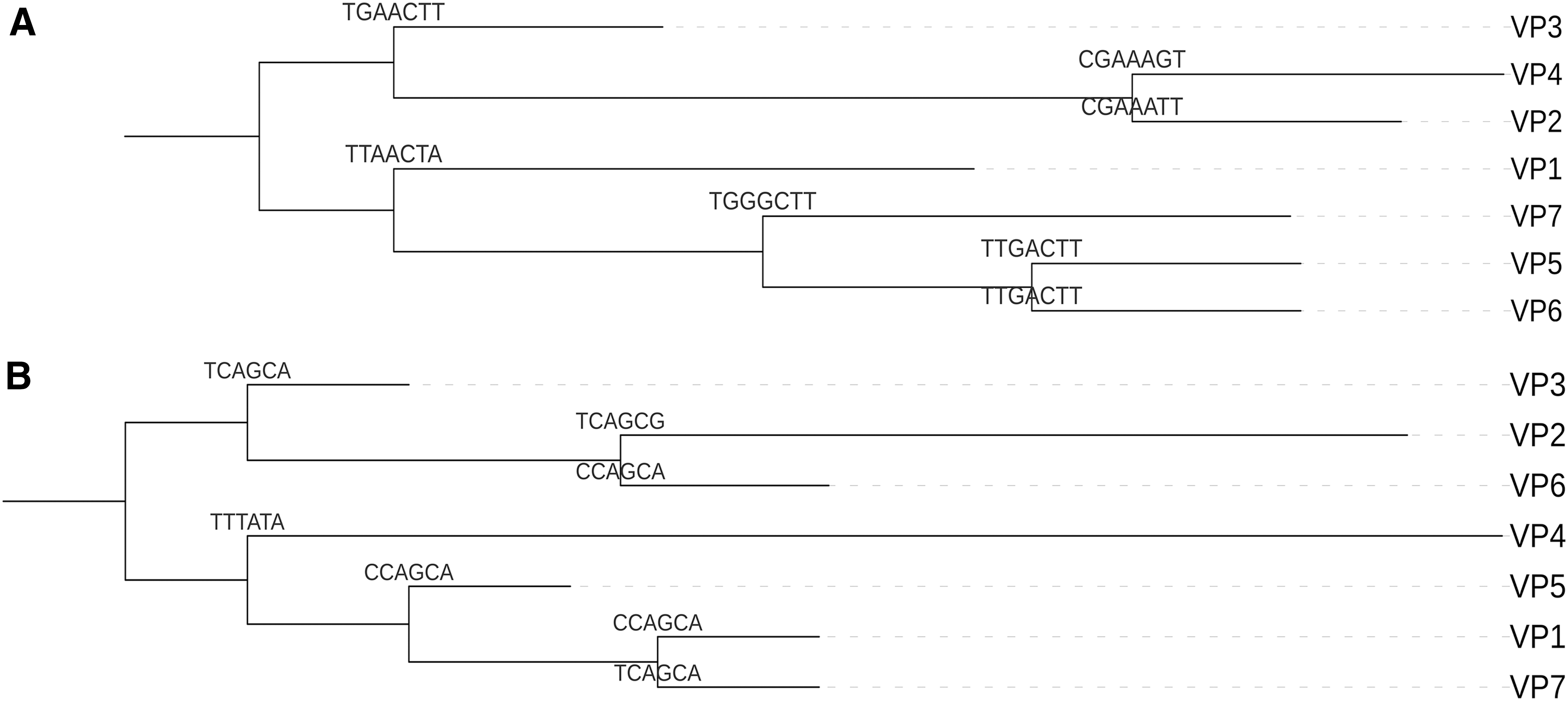
Phylogenetic trees of the VP isolates constructed based on SNPs found in chromosome 1
Single Nucleotide Polymorphisms Found in Chromosomes 1 and 2 of Each Vibrio parahaemolyticus Isolate, Their Respective Loci, Position in Base Pairs, Change in Nucleotide, Change in Amino Acid of the Respective Protein Product, and the Type of Single Nucleotide Polymorphism Variation
Very light gray, synonymous variant; light gray, missense variant; gray, noncoding transcript variant; dark gray, presence of single nucleotide polymorphism.
Comparative analysis of circular genomes
Figure 4 represents whole-genome comparison between the seven phenotypically identical isolates VP1-VP7, the reference genome RIMD 2210633, and two pre-1996 pandemic strains AQ3810 and AQ4037. Circular alignment of the seven collected genomes showed at least a 95% match with the representative pandemic clone, while several gaps were detected when compared with AQ3810 and AQ4037. This difference could be attributed to the additional genetic elements present in VP1-VP7 while missing in the pre-1996 pandemic strains.
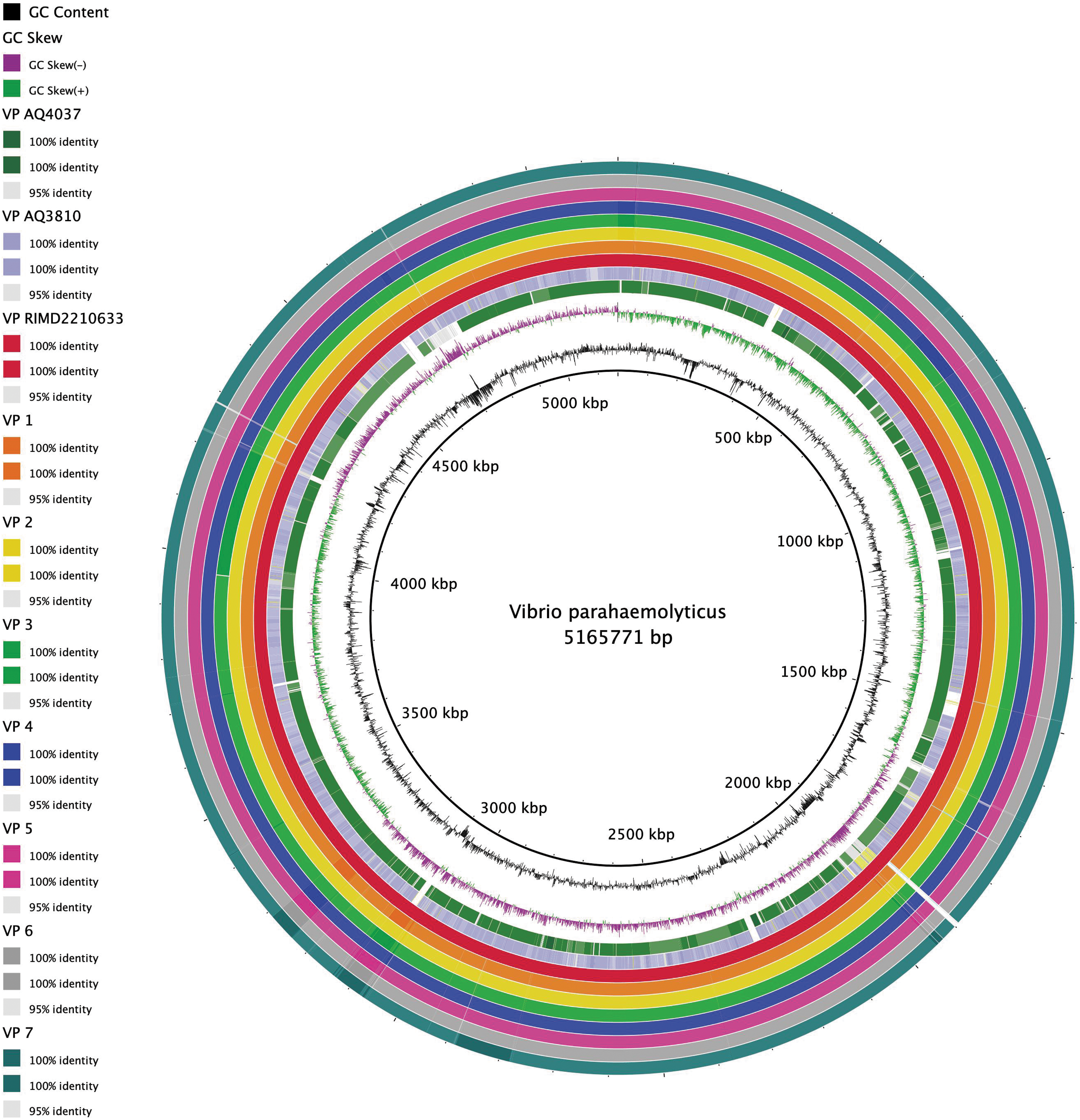
Comparative analysis of circular genomes containing both chromosomes of the VP isolates with three reference strains, RIMD 2210633, AQ3810, and AQ4037. This image was generated by BRIG 0.95 with a maximum and minimum identity threshold of 100% and 95%, respectively (Alikhan et al., 2011). The black lines represent the GC content, the green lines represent the positive GC skew, and the purple lines represent the negative GC skew. The innermost to outermost circular rings represent the following genomes: AQ4037, AQ310, RIMD 2210633, and VP1 to VP7. This figure shows that the seven isolates are at least 95% similar to RIMD 2210633. The gaps in AQ4037 and AQ3810 represent sequences not found in the other genomes, which is consistent with the fact that AQ4037 and AQ3810 lack pathogenicity islands found in RIMD 2210633, as well as one of the T6SSs in AQ3810. GC, guanine-cytosine; T6SS, Type VI secretion system; VP, Vibrio parahaemolyticus.
Discussion
V. parahaemolyticus outbreaks have been reported worldwide in countries such as Japan, China, Spain, France, Italy, Mexico, and the United States (Chen et al., 2016; Martinez-Urtaza et al., 2016; Han et al., 2017). Although it remains unclear if the gain of the orf8 from the vibriophage f237 has contributed to the widespread distribution of the pandemic strain O3:K6, it has provided a suitable genetic marker for the detection of this serotype (Nasu et al., 2000; Hazen et al., 2015; Wang et al., 2017).
In this study, a number of isolates recovered from patients suffering from gastroenteritis during summer 2017, were collected from stool samples, identified and characterized through WGS and molecular typing. Results obtained confirmed that the isolates were associated with an outbreak caused by a V. parahaemolyticus clone ST3, which was previously associated with outbreaks worldwide (González-Escalona et al., 2008). Further analysis also revealed the presence of orf8, linking the isolates to the well-known pandemic O3:K6 serotype.
Pandemic O3:K6 strains have the tdh
+, trh
The toxin tlh is a species-specific marker, as well as a hemolysin, and encodes a phospholipase A2 (Federici et al., 2018). Meanwhile, the noncoding fragment pR72H is another important marker for V. parahaemolyticus, found on chromosome 1, and detected in both clinical and environmental isolates (Coutard et al., 2005; Federici et al., 2018). The amplification of this fragment was developed as a method to detect environmental V. parahaemolyticus strains, found in seafood, since those isolates do not have the species-specific hemolysin tlh (Lee et al., 1995). All the tested isolates were positive for tlh and pR72H, which additionally verified the species.
Vibrio species, in general, are susceptible to most antimicrobial agents (Han et al., 2007). However, V. parahaemolyticus has intrinsic resistance to penicillin (Chiou et al., 2015; Li et al., 2016). This was also noted in our results, where resistance to ampicillin (Table 1) along with the presence of VPA0477, a β-lactamase, specific for V. parahaemolyticus, were detected. Basic Local Alignment Search Tool (BLAST) analysis of the β-lactamase sequence showed that it matched with CARB-22, a carbenicillin-hydrolyzing class A β-lactamase in V. parahaemolyticus RIMD 2210633 found on chromosome 2 (Chiou et al., 2015). The bla CARB genes were observed in a broad range of organisms, possibly due to mobile genetic elements. Carbenicillinases were also found on chromosome 2 of Vibrio cholerae (Petroni et al., 2004). The other β-lactamases, bla SHV, bla OXA, and bla TEM, were not detected in this study.
The chromosomal VPA0095 gene leading to quinolone resistance in V. parahaemolyticus (Aedo et al., 2014), was not detected in all the isolates undertaken in this study. Intermediate resistance to ciprofloxacin, a fluoroquinolone, was observed in VP1.
All the isolates in this study had the same ST type, serotype, virulence genes, and resistance patterns. Additionally, the analysis of the PFGE results, which is a common typing approach, showed that the isolates belonged to one pulsotype, except for VP1 that was untypable (Fig. 1). Having untypable strains is considered as a major PFGE drawback when studying Vibrio species (Ellingsen et al., 2008). WGS analysis could help accordingly, in overcoming the low resolution of such typing approaches, and help in better elucidating the relatedness of isolates (Tsai et al., 2017).
Pangenome analysis using Roary was another approach that we used to compare and determine the differences between the seven isolates, especially that of VP1. The isolates were clustered based on the presence and absence of common genes (Fig. 2). Some of the 293 unshared protein products included the IS4 family transposase ISVsa5, the elongation factors Tu and Tu2, the sulfate adenylyltransferase subunit 1, the electron transport complex subunit RsxC, the sulfur carrier protein TusA, flagellin D, and the urease accessory protein UreE. However, only the presence of glnS, encoding a rare cytosolic glutamine-tRNA ligase, and pinR, encoding a serine recombinase, were unique to VP1. Moreover, a block of hypothetical proteins found between the 93 and 94 kb region was unique to VP3 and VP6 (Fig. 2B).
Furthermore, wgSNP-based trees were constructed for chromosome 1 (Fig. 3A) and chromosome 2 (Fig. 3B) separately. Results obtained showed that there were 3 synonymous and 3 missense variants in chromosome 1, whereas there were 5 synonymous, 1 missense, and 11 noncoding transcript variants in chromosome 2 (Table 2). These results also indicated a higher mutation rate in chromosome 2 compared with chromosome 1.
In contrast to the limited resolution of traditional molecular typing methods, such as PFGE and PCR-based assays, an end-to-end whole-genome sequence-based analysis helped us to differentiate between the closely related isolates; we were able to construct high-resolution phylogenetic trees based on variants of both core genome single nucleotides and key protein-coding regions.
Comparative circular genomes were generated using WGS data. VP1-VP7 showed at least 95% similarity when compared with the reference strain RIMD 2210633 (Fig. 4). The genome RIMD 2210633 is known to contain two circular chromosomes with seven integrated pathogenicity islands (PAI): VPaI-1 (VP0380-VP0403), VPaI-2 (VP0635-VP0643), VPaI-3 (VP1071-VP1094), VPaI-4 (VP2131-VP2144), VPaI-5 (VP2900-VP2910), VPaI-6 (VPA1253-VPA1270), and VPaI-7 (VPA1312-VPA1398) (Chi and Wong, 2017).
Alignment and comparison of known genes in the different PAIs within the studied isolates and that of RIMD 2210633 showed that all had the seven PAIs. VPaI-1 contained a type I restriction endonuclease, VPaI-2 had a ribonuclease H1 protein, VPaI-3 encoded a methyl-accepting chemotaxis protein, VPaI-4 included the M protein, VPaI-5 contained two integrases, and VPaI-6 had two putative colicin proteins (Hurley et al., 2006). VPaI-7, found on chromosome 2, is the most studied island and contains two tdh genes and a T3SS, a cytotoxic necrotizing factor, an exoenzyme T gene, and five transposase encoding genes (Makino et al., 2003). The presence of the seven PAIs, in addition to the genotypes of the recovered isolates, additionally verified that they all had the same characteristics as that of the current pandemic clone.
However, BLAST analysis of the sequences found in the gaps shared by the seven isolates showed that they corresponded to genes encoding oxidoreductase, aldehyde dehydrogenase, DNA-binding transcriptional repressor, glutamine amidotransferase, hypothetical proteins, and glutamine synthetase in the 1900 kbp region. Moreover, the presence of gaps in the AQ3810 sequence revealed that AQ3810 had missing genomic elements. BLAST analysis of the sequences showed that it corresponded to VPAI-1 to VPAI-6. Figure 4 showed a gap associated with a high guanine-cytosine (GC) content; a characteristic feature distinguishing PAIs. This goes hand in hand with the fact that AQ3810 had six missing PAIs in its genome (Boyd et al., 2008). Other gaps found in the AQ3810 sequence were due to the two missing T6SSs that are present in RIMD 2210633 (Boyd et al., 2008).
In addition, BLAST analysis of the sequences representing the gaps found in AQ4037 indicated the absence of VPAI-7 and the two copies of tdh gene, present in RIMD 2210633.
Conclusion
To our knowledge, this is the first proven V. parahaemolyticus linked outbreak in Lebanon, as well as in the Middle East caused by ST3 O3:K6 serotype clone. Previous V. parahaemolyticus-related studies in Lebanon only showed the aquatic nature of the organism and how it is part of the microbiota of aquatic animals. The ability of the O3:K6 strain to spread on a global scale is concerning, especially for a bacterium that is not usually pathogenic. The identification of the tdh
+, trh
The lack of epidemiological data, as is the case in this study, is often a limitation in outbreak investigations. More studies are needed in the future to build a genome-based comparison between clinical and environmental isolates to determine the origin and source of contamination.
Footnotes
Acknowledgments
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.