
Abstract
A repetitive sequence-based polymerase chain reaction (rep-PCR) technique utilizing a semiautomated system, namely DiversiLab, was applied to determine the genotypes of Staphylococcus aureus and Bacillus cereus obtained from slaughterhouses. Twenty-four S. aureus and 16 B. cereus isolates from pigs and Hanwoo cattle from three slaughterhouses were used to create a DNA fingerprint library with the system software. Scatterplots demonstrated that rep-PCR groupings of S. aureus isolates were in good agreement with their origins. Specifically, linked rep-PCR profiles were observed for S. aureus isolates recovered from the same slaughterhouse, and higher genetic similarities were found among strains isolated from adjacent regions. All S. aureus isolates except one (ID: A-Hanwoo-9) from slaughterhouse “A” clustered with the three S. aureus reference strains, Korea Culture Center of Microorganisms (KCCM) 41291, KCCM 12214, and Culture Collection of Antimicrobial Resistant Microbes (CCARM) 3A007 (similarity values >95%). Moreover, most isolates obtained from slaughterhouse “B” clustered with S. aureus KCCM 11335 and KCCM 41331, and two isolates from slaughterhouse “C” clustered with CCARM 0027. Therefore, for this species, genotypic characteristics of regional isolates can be used to track the pathway of contamination. In contrast, B. cereus isolates showed high genetic diversity and could not be clustered with any specific group. Collectively, this system is useful for analyzing genetic diversity and is a rapid and reproducible typing method; however, there is a need to develop rep-PCR libraries for its use as a rapid identification method.
Introduction
Foodborne pathogenic bacterial diseases are a serious concern for public health in the food industry (Manoj et al., 2015). The consumption of contaminated pork and beef is a major source of human infection (Van Loock et al., 2000) and the microbial hazards associated with meat originate from poor hygiene and processing conditions in the slaughter plant. During slaughter, many steps related to processing can contribute to the cross-contamination of carcasses with microorganisms, which originate from the animal hide/skin, processing equipment, workers, and most importantly, the gastrointestinal tracts of the animals (Borch and Arinder, 2002). Different molecular typing reports on foodborne pathogens from the environment and products of meat-processing plants indicate the presence of a limited number of strains or genotypes, together with the persistence of specific strains, even after cleaning and disinfection procedures (De Busser et al., 2011). Specifically, isolates recovered from the final products have been shown to belong to the same genotype as those isolated from processing equipment (Lunden et al., 2003); moreover, some strains were found to be unique to a particular region (De Busser et al., 2011). Therefore, the genotyping of foodborne pathogens is epidemiologically important to determine the source of contamination and the relationships among strains isolated from neighboring locations.
Staphylococcus aureus and Bacillus cereus are important foodborne pathogenic bacteria found in food products of animal origin (Gueven et al., 2006; Sakwinska et al., 2009). These organisms have been examined by a wide array of techniques, including pulsed-field gel electrophoresis (PFGE), restriction fragment length polymorphism, and random amplified polymorphic DNA (RAPD) polymerase chain reaction (PCR) (Nikbakht et al., 2008; Saei et al., 2010; Thorsen et al., 2010; Merzougui et al., 2014). Although these techniques are accurate and have good discriminatory power, they have not been standardized in terms of consistency and reliability (Healy et al., 2005) and often have a turnaround time of days or weeks (Wise et al., 2009). Consequently, a reliable and rapid typing method is needed to identify potential sources of contamination and control these pathogens in the environment. Recently, repetitive sequence-based PCR (rep-PCR) became commercially available in an automated format known as the DiversiLab system, which produces data that are more standardized and reproducible than those produced by manual, gel-based rep-PCR analyses (Healy et al., 2005). Basically, rep-PCR uses primers that target noncoding, naturally occurring repetitive DNA sequences interspersed throughout bacterial and fungal genomes (Versalovic et al., 1994). Bacterial delineation or substrain discrimination can then be achieved by electrophoresis when amplified DNA fragments or amplicons comprise a genomic fingerprint of different band sizes (Versalovic and Lupski, 2002). The DiversiLab system has been successfully used to characterize many microorganisms at the strain level, including Salmonella spp. (Wise et al., 2009), Escherichia coli (Chon et al., 2012), S. aureus (Shutt et al., 2005), B. cereus (Kim et al., 2016), and Listeria monocytogenes (Abay et al., 2012).
The aims of this study were to investigate the genotypes of S. aureus and B. cereus from pigs and Hanwoo cows (a Korean cattle breed) from slaughterhouses in various parts of South Korea using the DiversiLab automated rep-PCR system, and to identify contamination routes and molecular diversity.
Materials and Methods
Reference strains and culture
Eleven B. cereus reference strains (Culture Collection of Antimicrobial Resistant Microbes [CCARM] 0002, CCARM 0120, CCARM 0136, Korea Culture Center of Microorganisms [KCCM] 11341, KCCM 11774, KCCM 12142, KCCM 40022, KCCM 40133, KCCM 40152, KCCM 40154, and American Type Culture Collection [ATCC] 11778) and 13 S. aureus reference strains (CCARM 3A007, CCARM 3A011, CCARM 3A220, CCARM 0027, KCCM 11335, KCCM 11640, KCCM 12214, KCCM 12255, KCCM 41291, KCCM 41294, KCCM 41324, KCCM 41331, and KCCM 41446) were obtained from the KCCM and the CCARM in Korean, and the ATCC in United States of America to construct the initial pathogen libraries. Bacteria were cultured in tryptic soy broth (BD Difco) at 37°C for 18 h.
Sample perpetration and isolation
All samples (103 samples from pigs and 103 samples from Hanwoo cattle, including environmental samples from the slaughtering process) were obtained from three slaughterhouses located in different parts of Korea. In this study, the three slaughterhouses are referred to as “A,” “B,” and “C.” The samples included environmental samples (from the surfaces of slaughterhouse equipment, and water and feed boxes for livestock), carcasses during the slaughtering process, and organ samples (intestine, stomach, and lung). The environmental samples were collected by swabbing areas of ∼100 cm2, and the animal carcasses and organ samples (25 g each) were homogenized in 225 mL of phosphate-buffered saline (pH 7.6) for 5 min in a Stomacher Lab-Blender (bioMérieux, Lyon, France) and incubated for 24 h at 30°C. Serial 10-fold dilutions of Stomacher fluids were then spread onto Baird Parker agar (BD Difco) or mannitol egg yolk polymyxin agar (BD Difco) plates and incubated at 37°C for 48 h.
Identification of isolates
Isolates were examined by Gram staining and catalase testing. Gram-positive and catalase-positive isolates were further identified using the API Staph and API CHB Kit (bioMérieux), as well as 16S rRNA sequencing analysis. The DNA of the isolates was extracted using the UltraCleanTM Microbial DNA Isolation Kit (Mo Bio Laboratories, Solana Beach, CA) following the manufacturer's instructions. PCR was performed using the universal primers, 27F and 1492R, as described by Chopra et al. (2014). DNA sequencing was performed with the ABI-Prism Big Dye Determinator Cycle Sequencing Ready Reaction Kit and ABI-Prism 377 Sequencer (Applied Biosystems Japan, Tokyo, Japan).
DiversiLab semiautomated rep-PCR DNA fingerprinting
The rep-PCR assay was performed using the DiversiLabTM Staphylococcus Kit or Bacillus Kit (bioMérieux) for DNA fingerprinting according to the manufacturer's instructions. Briefly, 2 μL of genomic DNA (final concentration, 25–50 ng/μL), 0.5 μL (or 2.5 U) of AmpliTaq® polymerase (Applied Biosystems, Foster City, CA), 2 μL of kit-supplied primer mix, and 2.5 μL of 10 × PCR buffer (Applied Biosystems) were added to 18 μL of kit-supplied rep-PCR master mix (MM1) for a total reaction volume of 25 μL per PCR. Thermal cycling parameters were as follows: initial denaturation at 94°C for 2 min; 35 cycles of denaturation at 94°C for 30 s, annealing at 45°C for 30 s, and extension at 70°C for 90 s; a final extension at 70°C for 3 min. The rep-PCR products were detected and separated using microfluidic chips and analyzed using the DiversiLab system.
Diversity analysis and library construction
Reference cultures were first characterized using the DiversiLab system software to identify the initial subspecies of the two pathogens (S. aureus and B. cereus) tested in this study. The DNA fingerprint patterns generated by the DiversiLab software (version 3.3) were viewed as both electropherograms and virtual gels. These samples were then compared with the library, and dendrograms with similarity matrices were calculated using the Kullback–Leibler distance correlation coefficient.
Results and Discussion
Isolation of Staphylococcus spp. and Bacillus spp.
The Staphylococcus spp. and Bacillus spp. isolates (38 and 31, respectively) were obtained from pigs and Hanwoo cattle from three different slaughterhouses, and their origins are listed in Table 1.
Occurrence and Distribution of Staphylococcus spp. and Bacillus spp. Isolated from Different Slaughterhouses
Staphylococcus spp. was most frequently isolated from the carcasses and skin of pigs and environmental samples from slaughterhouse “B,” followed by pig stomach and abdominal tissues from slaughterhouse “A”; only one Staphylococcus spp. strain, from pig lung, was detected in slaughterhouse “C.” The overall prevalence of Staphylococcus spp. in Hanwoo cattle was very low. Seven isolates were detected from slaughterhouses “A” and “C,” suggesting that the safety management for Hanwoo cattle slaughter was better than that for pigs in Korea. There is little data available regarding the prevalence of Staphylococcus during the slaughter of pigs and Hanwoo in Korea. However, Saide-Albornoz et al. (1995) investigated the occurrence of pathogens on pig carcasses at a specific point during the slaughter process and reported that S. aureus was the most prevalent pathogen, showing high levels of contamination. Many recent cases of methicillin-resistant S. aureus (MRSA) have also been associated with contaminated livestock, and particularly pigs, pig farms, and farmers (Van Duijkeren et al., 2007; Fluit, 2012). This suggests that the potential risk of foodborne diseases is not restricted to animals and associated environments; humans are also at risk due to the exchange of virulence factor-encoding genes between animal and human-associated strains (Graveland et al., 2010).
Bacillus spp. was extensively detected (19/31 isolates) in Hanwoo cattle of slaughterhouse “A,” including the stomach, small intestine, large intestine, outer abdominal wall, and lung (Table 1). In contrast, Bacillus spp. was detected in pigs only at specific sites in slaughterhouses, specifically an environmental sample of slaughterhouse “A,” carcasses in slaughterhouse “B,” and lung samples from slaughterhouse “C.” Therefore, we assume that Bacillus spp. isolates originate from not only pigs and Hanwoo cattle themselves but also from the environment of slaughterhouses due to the ubiquitous nature of Bacillus spp. in the environment. Limited data are available regarding the isolation of B. cereus during the slaughter process, although a small number of studies have assessed the prevalence of B. cereus contamination in cereal-based foods, almonds, and nuts (European Food Safety Authority [EFSA] and European Center for Disease Prevention and Control [ECDC], 2016). Recently, Choi et al. (2013) investigated the incidence and levels of microbial contamination in porcine carcasses and fresh pork from slaughterhouses and processing lines, and found that B. cereus was the most frequently detected pathogen in slaughterhouses. Furthermore, Gueven et al. (2006) determined that 22.4% of retail samples (of a total of 100), including meat and meat products, contained B. cereus.
Genotyping of S. aureus using automated rep-PCR
Of 38 Staphylococcus spp., 24 (63%) were identified as S. aureus by 16S rRNA sequencing analysis (Fig. 1). The identified S. aureus isolates were subjected to automated rep-PCR with the DiversiLab system to recognize identical strains and discriminate dissimilar strains. At 95% similarity, five distinct clusters were detected (Fig. 1). Clusters I, II, and III had 16 isolates (No. 1–16) and consisted of 14 strains obtained from pigs of slaughterhouse “B,” two isolates from Hanwoo cattle of slaughterhouse “A” (No. 12), and one pig from slaughterhouse “C” (No. 13). Cluster IV and Cluster V consisted of isolates from slaughterhouses “C” and “A,” respectively.

Dendrogram analysis and virtual gel image of DiversiLab semiautomated rep-PCR fingerprint analysis of the Staphylococcus aureus isolates from pig and Hanwoo cattle slaughterhouses. rep-PCR, repetitive sequence-based polymerase chain reaction. Color images are available online.
A scatterplot was also generated to provide a spatial view of the relationships among S. aureus strains (Fig. 2). These were classified into three groups according to slaughterhouse location. Strains from slaughterhouse “B” had similarities of up to 66.9% and 79% compared with those of slaughterhouses “A” and “C,” respectively. Indeed, slaughterhouse “B” is geographically closer to slaughterhouse “C” than to slaughterhouse “A.” Furthermore, the Pig-32 S. aureus strain (No. 13), isolated from a pig lung from slaughterhouse “C,” and Hanwoo-9 S. aureus strain (No. 12), isolated from Hanwoo cattle small intestine from slaughterhouse “A,” exhibited high genetic similarity (>95%) with isolates obtained from pig carcasses of slaughterhouse “B.” These results indicate that slaughterhouse-specific genetic diversity might exist, whereas cross-contamination through workers or vehicles could occur between adjacent slaughterhouses. Moreover, in this study, the same S. aureus genotypes were identified among isolates from intestinal contents, environments, and carcasses within one slaughterhouse, suggesting that cross-contamination occurs frequently during the slaughtering process. This finding is consistent with that of Vancleef et al. (2010), who reported that the same strains of livestock-associated MRSA were found in various animals, livestock farmers, and retail meat within one pig slaughterhouse in the Netherlands. Regarding slaughterhouse “B,” two pig S. aureus strains (No. 8 and No. 9) isolated from environmental samples (feed and water boxes) had very different genotypes compared with those of five other strains (No. 10, No. 11, No. 14, No. 15, and No. 16) isolated from carcasses. This results suggests that different S. aureus strains are present in the environment and the pig itself.
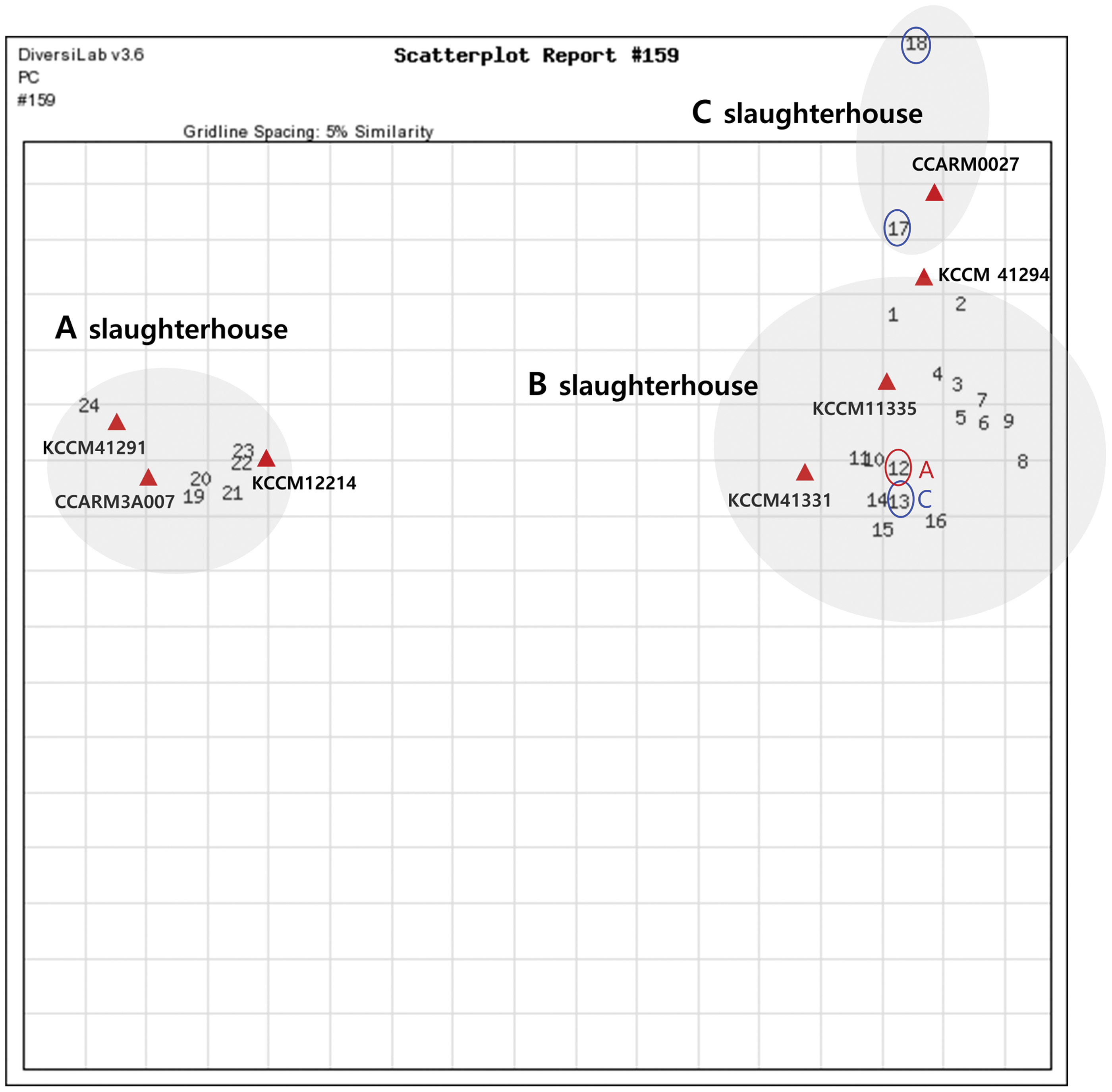
Scatterplot of S. aureus isolates and top match reference strains generated by rep-PCR using the DiversiLab System. The gridlines indicate an approximate percentage of similarity. rep-PCR, repetitive sequence-based polymerase chain reaction. Color images are available online.
Collectively, these results suggest that semiautomated rep-PCR is a useful tool to screen S. aureus isolates and track contamination in the food industry. However, several previous studies reported that PFGE is better than DiversiLab rep-PCR for accurately subtyping MRSA isolates (Babouee et al., 2011; Aguadero et al., 2015). However, a comparative evaluation showed that the performance of the DiversiLab system was comparable to that of PFGE for determining the strain relatedness of S. aureus isolates (Shutt et al., 2005; Grisold et al., 2010). Moreover, the semiautomated rep-PCR method has many advantages over PFGE in terms of reproducibility, standardization of the various steps, and the speed of execution (Pounder et al., 2006; Ben-Darif et al., 2010).
Genotyping of B. cereus using automated rep-PCR
A total of 16 isolates were identified as B. cereus by 16S rRNA sequencing. The dendrograms generated from rep-PCR patterns through the analysis of genotyping results of B. cereus isolates and computer-generated virtual gel images are shown in Figure 3. Furthermore, a scatterplot showing the genotypic proximity among B. cereus strains is presented in Figure 4. B. cereus showed a more diverse genotypic pattern than S. aureus, and this species was not clustered according to slaughterhouses. In addition, various genotypes were found within the same sample. For example, two B. cereus strains (No. 9 and No. 16) from pig lungs from slaughterhouse “C” had different genotypes and five strains (No. 5, No. 7, No. 8, No. 13, and No. 14) from pig carcasses in slaughterhouse “B” had three distinct genotypic patterns. The rep-PCR patterns of B. cereus isolates from the Hanwoo cattle slaughterhouse also varied, even though they were obtained from the same slaughterhouse. In particular, two strains (No. 12 and No. 15) isolated from the large intestine of Hanwoo cattle exhibited very low similarity (62.3%). This observation is consistent with observations reported by Oh et al. (2012), wherein B. cereus strains had very diverse genotypic patterns when analyzed by RAPD. Furthermore, an analysis of B. cereus isolated from food products marketed in Belgium also demonstrated very high diversity in terms of multilocus genotypes (Samapundo et al., 2011). These results indicate that the exchange of genetic material occurs frequently in the natural environment of B. cereus.
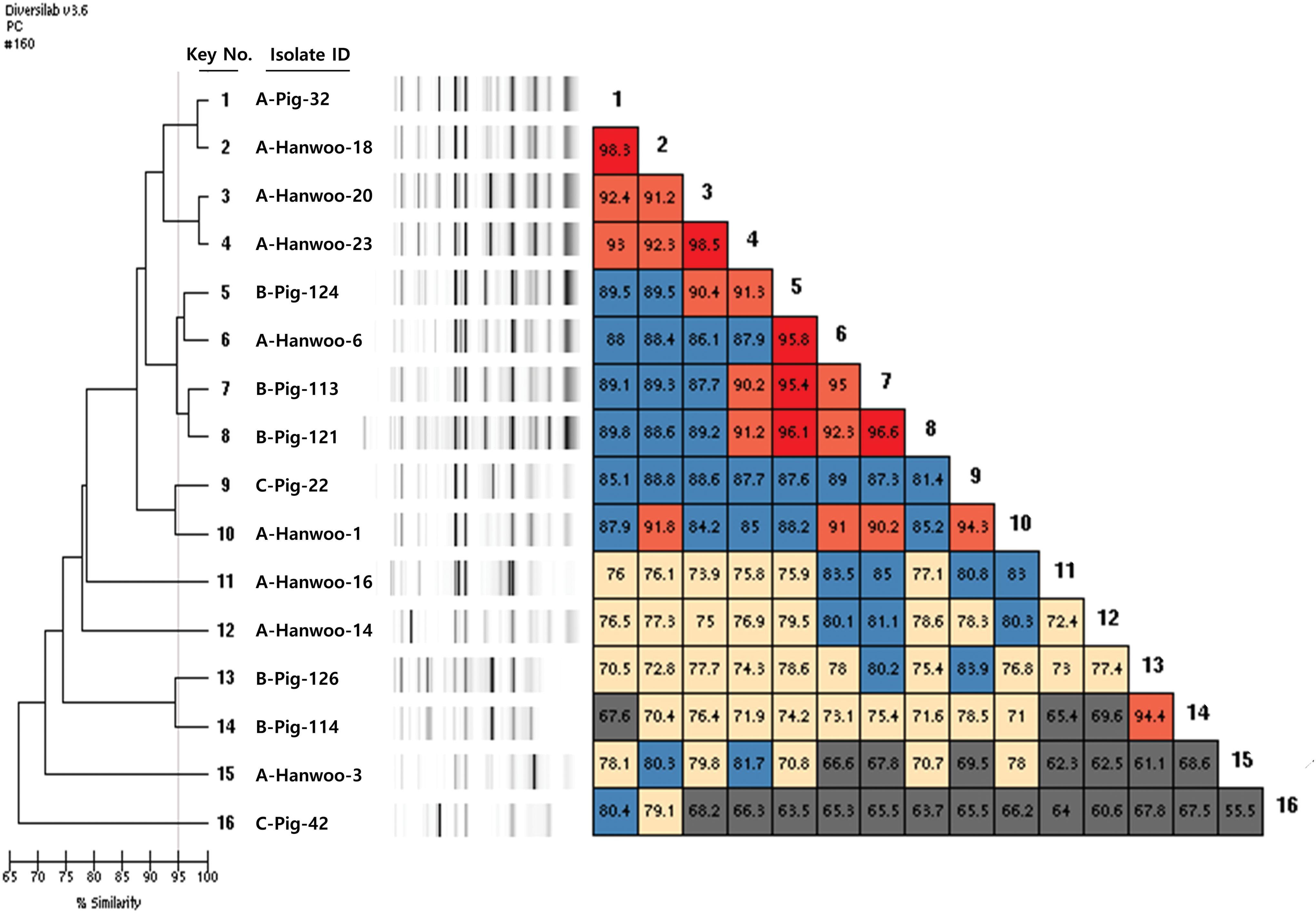
Dendrogram analysis and virtual gel image of DiversiLab semiautomated rep-PCR fingerprint analysis of the Bacillus cereus isolates from pig and Hanwoo cattle slaughterhouses. rep-PCR, repetitive sequence-based polymerase chain reaction. Color images are available online.
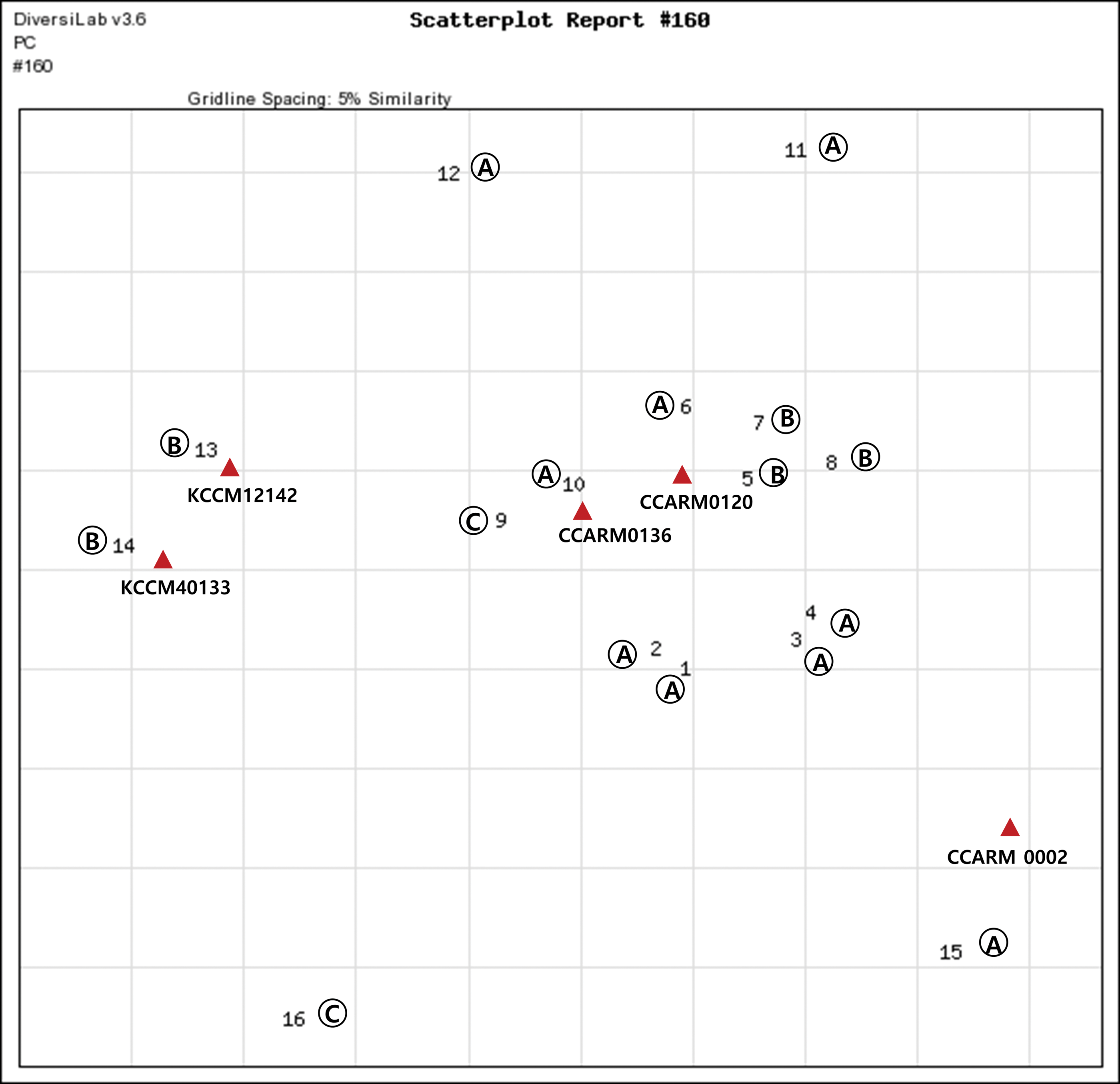
Scatterplot of B. cereus isolates and top match reference strains generated by rep-PCR using the DiversiLab System. The gridlines indicate an approximate percentage of similarity. rep-PCR, repetitive sequence-based polymerase chain reaction. Color images are available online.
Prediction of S. aureus and B. cereus through comparisons with the library
A total of 40 isolates identified as S. aureus or B. cereus were typed with the DiversiLab System and queried against the reference strain library. The clustering of the DiversiLab dendrogram and the Top match function of the DiversiLab classification report was subsequently utilized as a guide to predict genotypes.
Of the 24 S. aureus isolates, 15 were in accordance with the libraries with a >95% similarity threshold (Table 2). Especially, all isolates except one (No. 12) from slaughterhouse “A” exhibited more than 95% similarity to the three reference strains of S. aureus, namely KCCM 41291, KCCM 12214, and CCARM 3A007 (Fig. 2). Moreover, most of the isolates obtained from slaughterhouse “B” were clustered with S. aureus KCCM 11335 and KCCM 41331, and two isolates from slaughterhouse “C” were clustered with CCARM 0027. These results demonstrate that rep-PCR can be used to rapidly predict the genotype of S. aureus according to slaughterhouse location. However, caution is advised in employing the 95% threshold value for comparison with a preexisting library. Wise et al. (2009) reported that a query sample was 96.8% similar to the Salmonella serotype Kentucky based on automated rep-PCR with DiversiLab, but conventional serotyping returned the result of Lille serotype.
Identification of Staphylococcus aureus Isolates by Comparison with Automated Repetitive Sequence-Based Polymerase Chain Reaction Profiles
rep-PCR, repetitive sequence-based polymerase chain reaction.
Most (12/16 isolates) of the isolates identified as B. cereus by 16S rRNA sequencing did not group with any library entry at the 95% threshold level (Fig. 3). Furthermore, some B. cereus isolates fell within the 85% similarity index, (Table 3) and thus, rep-PCR was not able to identify these species. This finding suggests that the library within the DiversiLab software is insufficient to cover all isolates examined in this study. Many researchers have reported difficulties in discriminating B. cereus strains due to their high level of genetic diversity (Pirttijarvi et al., 1999; Ehling-Schulz et al., 2005).
Identification of Bacillus cereus Isolates by Comparison with Automated Repetitive Sequence-Based Polymerase Chain Reaction Profiles
rep-PCR, repetitive sequence-based polymerase chain reaction.
Although this system has many advantages such as fast assay times and high reproducibility, it might present some drawbacks if the reference data from the library are insufficient to analyze the target bacteria. Therefore, the typing results from the DiversiLab system should be reported with caution if the accumulated library data are inadequate or if no epidemiological data are available. Accordingly, the internal library should be continuously updated with new serotypes from various sources to facilitate the reliable application of this system.
Conclusions
The semiautomated rep-PCR-based method was used to determine the subtypes of S. aureus and B. cereus isolated from slaughterhouses. S. aureus rep-PCR patterns were in good agreement with the origins of these isolates, and linked rep-PCR profiles were observed for S. aureus isolates recovered from the same slaughterhouse. In addition, higher genetic similarities were observed among strains isolated from adjacent regions. Consequently, the genotypic characterization of regional isolates using the DiversiLab rep-PCR system can be used to track the pathway of contamination for this species. In contrast, B. cereus rep-PCR patterns were highly variable, which could be attributed to the higher diversity of B. cereus strains compared with that of S. aureus strains. The DiversiLab rep-PCR system might thus provide a rapid and standardized method for subtyping isolates and could be useful for predicting contamination pathways. However, for the system to be used to reliably identify species, future studies would be needed to expand the existing rep-PCR libraries with information from many reference strains.
Footnotes
Acknowledgments
This work was supported by the Cooperative Research Program for Agriculture Science and Technology Development (Project no. PJ007585201002) and the Postdoctoral Course Program of the National Institute of Animal Science, Rural Development Administration (Korea).
Disclosure Statement
No competing financial interests exist.