
Abstract
Klebsiella spp. is a common cause of clinical mastitis (CM) in dairy cows. However, relatively less information is available about distribution of virulence factors of Klebsiella spp. isolated from cows with CM. Objectives of this study were, therefore, to determine the prevalence of hypermucoviscosity (HMV) phenotype, capsule serotypes, and potential virulence genes in Klebsiella spp. from cows in China with CM. A total of 241 Klebsiella spp. isolates were recovered from cows with CM on 123 dairy farms (each had >500 lactating cows) located in 13 provinces of China. Of the isolates, 124 (51%) and 117 (49%) were identified as Klebsiella pneumoniae and Klebsiella oxytoca, respectively. The prevalence of HMV was 16% for K. pneumoniae and 11% for K. oxytoca; entB (78%), fimH1 (55%), kfu (31%), and mrkD (24%) were the prevalent virulence genes among K. pneumoniae, whereas entB (50%), fimH1 (30%), and mrkD (22%) were prevalent in K. oxytoca. Prevalence of the lac gene was higher for K. pneumoniae (78%) than for K. oxytoca (13%), whereas the nif gene was more prevalent in K. oxytoca than in K. pneumoniae (12% and 1%, respectively). Fifty-six K. pneumoniae isolates were confirmed as K57, the most prevalent capsule serotype (45%). Twenty-one (18%), 20 (10%), and 9 (8%) of 117 K. oxytoca isolates were positive for K57, K5, and K54 serotypes, respectively. As the predominant serotype, K. pneumoniae K57 isolates had a higher prevalence of the HMV phenotype and fimH1 than non-K57 K. pneumoniae. In conclusion, virulence factors were commonly detected for both K. oxytoca and K. pneumoniae causing CM in Chinese dairy herds. HMV isolates were commonly identified, irrespective of species. In addition, as the predominant capsule in bovine K. pneumoniae, the K57 serotype may be better adapted to the udder environment; therefore, further studies targeting pathogenicity to mammary tissue should contribute new knowledge for vaccine development using this serotype.
Introduction
Gram-negative bacteria, mostly coliforms (Escherichia coli, Klebsiella spp., and Enterobacter spp.), are important bacterial causes of mastitis, particularly in well-managed dairy herds. Klebsiella spp. have become common coliform bacteria causing clinical mastitis (CM) (Roberson et al., 2004; Olde Riekerink et al., 2008; Oliveira et al., 2013). In a recent Chinese study (Gao et al., 2017), Klebsiella spp. were isolated from 13% of CM samples of 61% of participating dairy farms, comparable in prevalence with E. coli (14% of samples and 75% of herds).
Although CM due to Klebsiella spp. is often mild to moderate with prolonged duration and substantial loss of milk yield (Grohn et al., 2004), it can also be severe with increased mortality rates. Virulence factors of Klebsiella spp. isolates may be a critical reason. Various factors, such as capsular serotypes (K1, K2, K5, K54, and K57), hypermucoviscosity (HMV), iron-scavenging systems (e.g., aerobactin, enterobactin, yersiniabactin, and phosphotransferase), fimbria-encoding systems (e.g., adhesin type 1 fimbriae encoded by the gene fimH1 and adhesin type 2 fimbriae encoded by mrkD), allantoin metabolism, and regulation of mucoid phenotype A, have been reported to be involved in virulence of Klebsiella spp. isolates of human origin (Jagnow and Clegg, 2003; Struve et al., 2008; Russo et al., 2014; Clegg and Murphy, 2016; Ko, 2017).
Although virulence of human isolates has been widely described, distribution of virulence factors was rarely reported in animal-source Klebsiella spp., including isolates from bovine mastitis (Osman et al., 2014). Preliminary analyses indicated that specific genes may be associated with virulence of Klebsiella pneumoniae isolates from bovine mastitis, such as genes involved in lactose metabolism (Holt et al., 2015). It is still unknown whether virulence mechanisms and associated genes are similar in isolates from bovine mastitis and human infections; an initial assessment would rely on prevalence estimates of potential virulence genes in mastitis isolates. It is therefore important to explore and establish the prevalence of virulence genes in bovine Klebsiella spp. isolates from various countries or regions, including China.
Objectives of the present study were to estimate and compare the prevalence of the HMV phenotype and potential virulence genes in Klebsiella spp. isolated from cows with CM on large Chinese dairy farms, according to bacterial species and capsule serotypes.
Materials and Methods
Statement of ethics
Approval was granted by the ethics committee of the College of Veterinary Medicine, China Agricultural University (CAU), Beijing, and the study was conducted according to standard ethical guidelines implemented at CAU. Milk samples from cows with CM were collected according to National Mastitis Council (NMC, 2017) standard procedures and with appropriate consultation with the dairy farm's owner or administration.
Herds, isolates, and species identification
A total of 261 CM isolates from 261 cows obtained from March 2014 to May 2017 were preliminarily identified as Klebsiella spp. by microbiological identification, as described (Gao et al., 2017), including colony characteristics on MacConkey agar, Gram staining, motility, indole, ornithine reactions, oxidase, and growth on triple sugar iron slants. In the present study, 156 isolates were from a previous study (March 2014 to September 2016) on the incidence of CM in large Chinese dairy herds (Gao et al., 2017), whereas the remaining 105 isolates were collected from October 2016 to May 2017. Each Klebsiella spp. strain was isolated from 1 milk sample collected from a CM case; thus, 261 isolates were from 261 CM quarter milk samples. Cases of CM were detected routinely by herdsmen at milking time and confirmed by a veterinarian. Herd veterinarians were asked to collect a milk sample aseptically from quarters that had visible signs of CM (abnormal milk and/or swollen quarter(s) before antibiotic treatment), following standardized protocols (NMC, 2017). Milk samples were commonly stored in a freezer on the farm (at approximately −20°C), then placed on an ice pack, and sent (express mail) to the Mastitis Diagnostic Laboratory of China Agricultural University (MDL-CAU, Beijing, P.R. China) for bacterial culture. In the present study, a herd was subjectively defined as large if the farm contained >500 lactating cows, as reported (Gao et al., 2017). Dairy herds that voluntarily participated in a mastitis control program were enrolled and followed for >3 years.
Assuming a population size of 2.5 million lactating dairy cows in China and a Klebsiella spp. CM incidence rate of 0.43 cases per 100 cow-months in China (Gao et al., 2017), 196 isolates would be required to estimate a proportion of 0.15 (e.g., 15% prevalence of a potential virulence gene in Klebsiella spp. isolated from bovine mastitis) with a precision of 0.05 (5%). Since isolates are clustered within herds, assuming an intraclass correlation of 0.3 (high) and an average of 2 isolates sampled per herd, 255 isolates collected from different herds would be necessary for prevalence estimation of potential virulence factors.
These suspected Klebsiella spp. isolates were identified to the species level using partial 16S rDNA sequencing (Barry et al., 1990). A K. pneumoniae reference strain (ATCC 700603) was used as control.
Detection of HMV phenotype
Klebsiella spp. isolates were streaked on 5% sheep blood agar and incubated overnight at 37°C. They were classified as the HMV or non-HMV phenotype by the string test, in which a bacterial loop was used to stretch a string of mucous from the bacterial colony. Formation of a viscous string >5 mm was defined as positive (Fang et al., 2004).
Detection of capsular serotypes and virulence genes
Genomic DNA was extracted from overnight culture using a bacterial DNA extraction kit (TransGen, Beijing, China). PCR was used to detect potential virulence genes or operons (entB, aerobactin operon, ybtS, kfu, iutA, fimH-1, mrkD, allS, rmpA, lac, and nif) and capsular serotypes (K1, K2, K5, K54, and K57). Primers used for amplification and reference source are shown (Table 1). PCR primers for genes lac and nif were chosen from sequences (lac from accession number M11441 and nif from accession number X13303) in the GenBank database. Reaction mixtures (25 μL) consisted of 12.5 μL of Taq Mix (TransGen, Beijing, China), 1 μL of template DNA, 1 μL of each primer (10 μM; Sunbiotech, Beijing, China), and 9.5 μL of ultrapure distilled water. Initial denaturation at 95°C for 5 min was followed by 30 cycles of amplification at 95°C for 45 s, primer-specific annealing temperature for 30 s, extension at 72°C for 60–90 s, and a final extension step at 72°C for 10 min. PCR products were analyzed on a 2% agarose gel. For confirmation of lac and nif genes, 30 respective amplicons were randomly selected and sequenced. For other potential virulence genes, we confirmed that an isolate carried a target gene if the same size amplicon was observed, as reported (Fang et al., 2004, 2007; Yu et al., 2007; Turton et al., 2008; Compain et al., 2014; Wasfi et al., 2016), from which the primer sequences of those virulence genes were derived.
Primers Used in This Study
Statistical analyses
Statistical analyses were performed using R (R Core Team, 2017). Prevalence and 95% confidence interval (95% CI) estimation of potential virulence factors was done at the isolate level using generalized linear mixed models in R with herd random effects using the lme4 package (Bates et al., 2015). Models were estimated using maximum likelihood, with 30 quadrature points per scalar using the adaptive Gauss–Hermite quadrature. Prevalence of potential virulence factors and capsular serotypes was compared between K. pneumoniae and Klebsiella oxytoca using the same strategy, except for the rmpA gene and capsular serotype K1 where Fisher's exact test was used. Similarly, prevalence of potential virulence genes in K57 isolates and others was compared using generalized linear mixed models. p-Values obtained were adjusted for multiple comparisons by the Benjamini, Hochberg, and Yekutieli control of false discovery rate (Benjamini et al., 2001) using the p.adjust function in R. Statistical significance was set at 5%.
Results
Distribution of isolates and species identification
Twenty isolates confirmed as non-Klebsiella species were excluded from the study; the remaining 241 Klebsiella spp. isolates were from 123 dairy herds in 13 provinces (Fig. 1). On average, two isolates were obtained per farm: 59% of farms contributed one isolate/farm, 18% of farms contributed two isolates/farm, 11% of farms contributed three isolates/farm, 5% of farms contributed four isolates/farm, 3% of farms contributed five isolates/farm, 2% of farms contributed six isolates/farm, and 1%, 1%, and 1% of farms each contributed seven, eight, and nine isolates/farm. Among provinces contributing more than 10 Klebsiella spp. isolates (Inner Mongolia = 68, Hebei = 46, Heilongjiang = 22, Shandong = 17, Shanghai = 17, Anhui = 16, Liaoning = 15, Beijing = 13, and Henan = 12), K. pneumoniae was the prevalent species in Beijing (92%), Henan (67%), Anhui (63%), and Hebei (61%), whereas K. oxytoca was the prevalent species in Liaoning (80%) and Inner Mongolia (66%).

Geographic distribution of 123 large (>500 lactating cows) dairy herds in 13 provinces of China.
HMV phenotype of Klebsiella isolates
Hypermucoid characteristics of K. pneumoniae on blood agar and MacConkey agar are presented (Fig. 2). Among the 124 K. pneumoniae isolates, 31 (prevalence of 16%) isolates had the HMV phenotype, whereas 14 (prevalence of 11%) of 117 K. oxytoca isolates were hypermucoid (p = 0.06).
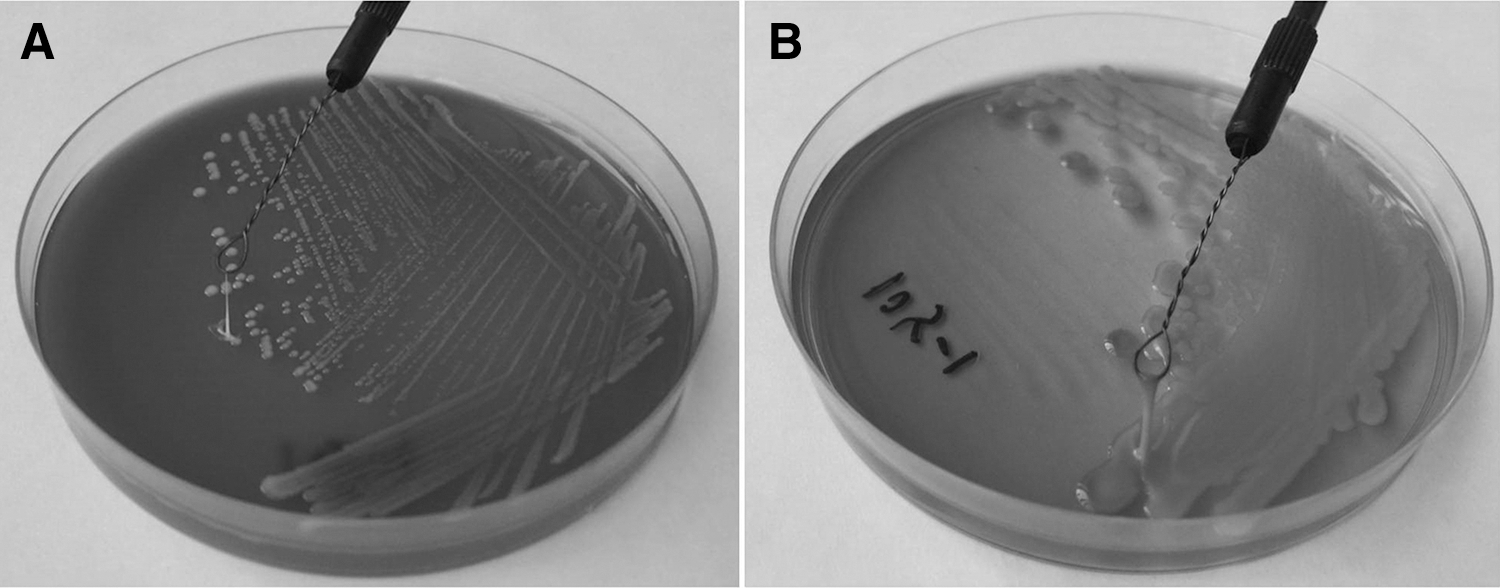
Hypermucoid characteristic of a Klebsiella pneumoniae strain. Formation of viscous string of the K. pneumoniae strain, >5 mm; therefore, T15-1 was defined as a hypermucoviscosity strain.
Prevalence of virulence genes in Klebsiella spp.
The most prevalent genes detected in K. pneumoniae isolates were entB and lac (prevalence of 78%), followed by fimH1(55%), kfu (31%), and mrkD (24%). The most prevalent gene in K. oxytoca was entB (50%), followed by fimH1 (30%), mrkD (22%), and kfu (14%; Table 2). A total of 177 (73%) Klebsiella isolates harbored at least one iron acquisition-related gene. entB, kfu, fimH1, rmpA, and lac were more frequently detected in K. pneumoniae, whereas the nif gene was more prevalent in K. oxytoca (Table 2).
Prevalence per 100 Isolates of Potential Virulence Factors Among Klebsiella spp. Isolates
Experimental conditions: Genomic DNA was extracted from overnight culture using a bacterial DNA extraction kit (TransGen, Beijing, China). PCR was used to detect potential virulence genes (entB, aerobactin operon, ybtS, kfu, iutA, fimH-1, mrkD, allS, rmpA, lac, and nif). The reaction mixture (25 μL) consisted of 12.5 μL of Taq Mix (TransGen, Beijing, China), 1 μL of template DNA, 1 μL of each primer (10 μM; Sunbiotech, Beijing, China), and 9.5 μL of ultrapure distilled water. Initial denaturation at 95°C for 5 min was followed by 30 cycles of amplification at 95°C for 45 s, primer-specific annealing temperature for 30 s, extension at 72°C for 60–90 s, and a final extension step at 72°C for 10 min. PCR products were analyzed on a 2% agarose gel.
Prevalence estimates were obtained using generalized linear mixed models with herd random effects.
p-Value comparing the prevalence of a potential virulence factor between Klebsiella pneumoniae and Klebsiella oxytoca adjusted for multiple comparisons (Benjamini et al., 2001).
Aerobactin operon.
95% CI, 95% confidence interval.
Capsular serotypes of Klebsiella spp. isolates
In the present study, K57 was the most prevalent capsular serotype in the 124 K. pneumoniae isolates (prevalence of 45%), followed by K1 (15%), K5 (10%), and K54 (6%) serotypes (Table 3). In K. oxytoca, K5 and K57 were the most prevalent serotypes (10% and 18%, respectively), followed by K54 (8%), whereas K1 was not detected in K. oxytoca (Table 3). K1 and K57 serotypes were more prevalent in K. pneumoniae than in K. oxytoca. K2 was not detected in any Klebsiella spp. isolate.
Prevalence per 100 Isolates of Capsule Serotypes Among Klebsiella spp. Isolates
Experimental conditions: Genomic DNA was extracted from overnight culture using a bacterial DNA extraction kit (TransGen, Beijing, China). PCR was used to detect capsular serotypes (Fang et al., 2004, 2007; Yu et al., 2007; Turton et al., 2008). The reaction mixture (25 μL) consisted of 12.5 μL of Taq Mix (TransGen, Beijing, China), 1 μL of template DNA, 1 μL of each primer (10 μM; Sunbiotech, Beijing, China), and 9.5 μL of ultrapure distilled water. Initial denaturation at 95°C for 5 min was followed by 30 cycles of amplification at 95°C for 45 s, primer-specific annealing temperature for 30 s, extension at 72°C for 60–90 s, and a final extension step at 72°C for 10 min. PCR products were analyzed on a 2% agarose gel.
Prevalence estimates were obtained using generalized linear mixed models with herd random effects.
p-Value comparing the prevalence of a capsule serotype between K. pneumoniae and K. oxytoca adjusted for multiple comparisons (Benjamini et al., 2001).
95% CI, 95% confidence interval.
Distribution of HMV phenotype and potential virulence genes was subsequently compared between K57 and non-K57 K. pneumoniae (Table 4). HMV isolates were more prevalent (38%) in K57 K. pneumoniae than in non-K57 isolates (5%). fimH1 was detected in higher proportions of K57 isolates (75%) than in non-K57 isolates (31%). In contrast, rmpA was detected in 25% of non-K57 isolates, but not in K57 isolates, as all 17 isolates carrying rmpA belonged to the K1 serotype. Distribution of the lac gene was not different between K57 and non-K57 K. pneumoniae (Table 4).
Prevalence per 100 Isolates of Virulence Genes in 56 K57 and 68 Non-K57 Capsule Serotypes of Klebsiella pneumoniae
Experimental conditions: For the HMV phenotype test, Klebsiella spp. isolates were streaked on 5% sheep blood agar and incubated overnight at 37°C. They were classified as HMV or non-HMV phenotype by the string test, in which a bacterial loop was used to stretch a string of mucous from the bacterial colony. Formation of a viscous string >5 mm was defined as a positive test (Fang et al., 2004). For virulence gene detection, genomic DNA was extracted from overnight culture using a bacterial DNA extraction kit (TransGen, Beijing, China). PCR was used to detect potential virulence genes (entB, aerobactin operon, ybtS, kfu, iutA, fimH-1, mrkD, allS, rmpA, lac, and nif) and capsular serotypes. The reaction mixture (25 μL) consisted of 12.5 μL of Taq Mix (TransGen, Beijing, China), 1 μL of template DNA, 1 μL of each primer (10 μM; Sunbiotech, Beijing, China), and 9.5 μL of ultrapure distilled water. Initial denaturation at 95°C for 5 min was followed by 30 cycles of amplification at 95°C for 45 s, primer-specific annealing temperature for 30 s, extension at 72°C for 60–90 s, and a final extension step at 72°C for 10 min. PCR products were analyzed on a 2% agarose gel.
Prevalence estimates were obtained using generalized linear mixed models with herd random effects.
p-Value comparing the prevalence of a potential virulence factor between K. pneumoniae capsular serotype K57 and K. pneumoniae capsular serotype other than K57 adjusted for multiple comparisons (Benjamini et al., 2001).
Aerobactin operon.
95% CI, 95% confidence interval; HMV, hypermucoviscosity.
Discussion
Among Klebsiella spp. isolates, proportions of K. pneumoniae (51%) and K. oxytoca (49%) were similar to that reported in previously investigated farms (Munoz et al., 2008), but not in accordance with another report that K. pneumoniae was the most common species (Roberson et al., 2004). Zadoks et al. (2011) closely traced sources of K. pneumoniae and K. oxytoca inside and outside farms and reported that the isolation rate of K. pneumoniae was very high in samples from rumens, feces, and alleyways, whereas K. oxytoca was more commonly isolated from soil used to grow corn or alfalfa. Perhaps the cow's teat end or udder skin could be mainly contaminated with K. pneumoniae if the cow lies on the bedding with lot of feces (great amount of K. pneumoniae) or with K. oxytoca if the cow lies on fresh bedding such as chopped cornstalks or sawdust (great amount of K. oxytoca from soil in the field), thereby increasing the risk of these Klebsiella species entering into the udder. The high isolation rate of a single species (only K. pneumoniae or K. oxytoca) from a herd and the finding that the majority of herds contributed only one species indicate that mastitis caused by various Klebsiella species may be associated with different herd-level factors (e.g., factors related to increased teat-end exposure to feces for mastitis outbreaks caused by K. pneumoniae) in dairy herds, in agreement with previous studies (Zadoks et al., 2011; Langoni et al., 2015). Future studies to explore sources and transmission of K. pneumoniae and K. oxytoca in Chinese farms will be of great importance.
In K. pneumoniae of human origin, isolates displaying the HMV phenotype were considered more virulent than non-HMV strains (Shon et al., 2013). In recent years, HMV K. pneumoniae has also been isolated from various animals (Twenhafel et al., 2008; Jang et al., 2010; Osman et al., 2014; Davies et al., 2016). Although currently there is no evidence for an association between HMV and severity of bovine mastitis, the relatively high prevalence of HMV K. pneumoniae from cows with bovine CM warrants comparison of pathogenicity between HMV and non-HMV K. pneumoniae in a future study. Additionally, HMV K. oxytoca was apparently reported for the first time in the current study.
Most Klebsiella spp. isolates carried at least one iron acquisition-related gene, indicating that capability of acquiring iron may be an important factor for growth and proliferation of Klebsiella species in udders. The detection rate of entB in K. pneumoniae was similar to that from human isolates, whereas aerobactin operon and kfu were lower (Wasfi et al., 2016). In addition, there was lower prevalence of aerobactin operon and kfu in this study compared with other studies targeting bovine mastitis (Osman et al., 2014). The prevalence of fimbria genes (fimH1 and mrkD) detected in this study was lower than that in previous reports on isolates from humans, which reported that fimH1 and mrkD existed in nearly all K. pneumoniae strains (Ranjbar et al., 2016; Wasfi et al., 2016). Therefore, virulence factors from human Klebsiella spp. isolates cannot necessarily be interpreted as virulence factors in the context of bovine CM. The lac operon was reported to be associated with K. pneumoniae isolates from bovine mastitis (Holt et al., 2015). Although lower than that report, the lac gene was still detected in considerable K. pneumoniae isolates. In contrast, the low carriage rate of the nif gene was also in accordance with previous results (Holt et al., 2015), suggesting less importance of this nitrogen-fixing gene for K. pneumoniae invading the udder. Although a large difference in prevalence of these two operons was present in K. pneumoniae, no similar difference was detected for K. oxytoca.
Some capsular types in K. pneumoniae of human source, particularly K1, K2, K5, K54, and K57, are considered isolates with high virulence (Ko, 2017). The capsular serotype results in this study were not in accordance with another study, which reported that K1 and K2 serotypes were prevalent in K. pneumoniae from bovine mastitis (Osman et al., 2014). For HMV isolates, capsular serotypes K1, K2, K5, K16, K20, K54, and K57 and a new capsular serotype (KN1) have been described (Shon et al., 2013). HMV phenotypes were reported to be highly associated with the K1 serotype in K. pneumoniae from human sources (Yu et al., 2008). In the present study, HMV isolates were more frequently detected in K57 than in non-K57 isolates, indicating an association between these two virulence factors. Unfortunately, little is known about the prevalence of the HMV phenotype in K57 K. pneumoniae derived from human sources, limiting further comparisons. Based on high prevalence of the K57 serotype in K. pneumoniae from bovine CM, we inferred that this serotype should be studied further regarding adaptation within the udder, pathogenicity to mammary tissue, and vaccine development potential.
Although the HMV phenotype, capsular types, and some potential virulence genes in Klebsiella spp. isolates from bovine CM were detected, this study had some limitations, including lack of virulence data (e.g., severity of CM cases) to associate with detected characteristics and low sample size to accurately estimate the prevalence of uncommon virulence genes according to the capsular serotype. Additionally, we had to target virulence genes mainly from the literature of Klebsiella spp. isolated from humans. Anyway, the present study reported nationwide, primary epidemiological information on potential virulence factors among Klebsiella spp. isolates from bovine CM. Our research team is now exploring the relationship between phenotypic characteristics of bovine mastitis (e.g., clinical signs, severity, and treating duration) and virulent characteristics of Klebsiella spp. strains at the herd level. We are also conducting the cell infection model by Klebsiella strains to demonstrate that some special virulence factors (e.g., mucoid capacity, lactose metabolism capacity, and K57 capsule serotypes) could be crucial to increase pathogenicity of Klebsiella strains on mammary epithelial cells (e.g., injury and apoptosis of cells and related inflammatory responses). All these data will deeply clarify the virulent characteristics of Klebsiella spp. on bovine mastitis.
Conclusions
In Klebsiella spp. isolates from cows with CM in large Chinese herds, entB, lac, and fimH1 were the most prevalent potential virulence genes in K. pneumoniae, whereas entB was prevalent in K. oxytoca. The detection rate of entB in mastitis isolates was similar to that from human isolates. The high prevalence of K57, a serotype that displayed the HMV phenotype more frequently, indicated that this serotype had increased adaptability to the udder and greater potential to cause disease. Further studies involving this serotype are needed to better elucidate pathogenicity of mastitis caused by K. pneumoniae and the potential for vaccine development.
Footnotes
Acknowledgment
The authors thank Dr. John Kastelic (University of Calgary, Calgary, AB, Canada) for editing the manuscript.
Disclosure Statement
No competing financial interests exist.
Funding Information
This research was financially supported by the Beijing Municipal Natural Science Foundation (No. 6192013), the National Natural Science Foundation of China (No. 31772813, 31572587), the High-end Foreign Experts Recruitment Program (No. GDT20171100013), and the National Key R&D Project (No. 2016YFD0501203).