
Abstract
Listeria monocytogenes is a deadly foodborne pathogen, and infections can result in meningoencephalitis and sepsis with mortality rates of up to 30%. In this study, we performed comparative whole-genome analysis of 30 clinical isolates sequenced together with 32 previously sequenced clinical and food isolates from China. The data indicate that L. monocytogenes isolates belonging to the clonal complexes (CC) −1, −8, −9, −87, −121, and −155 are present in human clinical cases. The majority of isolates are from CC-87, 9, and 8 and overlap with those CCs previously reported on the basis of multilocus sequence typing for isolates from Chinese food products. Detailed genome analysis of isolates, representative of CCs in clinical and food products, revealed strong similarities both in their core- and accessory genomes indicating that they are highly related. When compared to genome sequences of isolates of a given CC worldwide, clinical isolates of China were distinct and clustered in unified clades. Our data indicate that epidemic clones of L. monocytogenes (CC-87, 9, and 8) with unusually high occurrence of plasmids are unique to China and suggest that common populations of L. monocytogenes clones are present in both clinical and food products in China.
Introduction
Listeria monocytogenes is a foodborne pathogen and the etiological agent of listeriosis, a severe systemic disease of animals and humans associated with very high mortality rates (Buchanan et al., 2017). The average prevalence of L. monocytogenes in food products in China has been estimated to be around 4.4% (Li et al., 2018). A case-fatality of >25% has been reported for invasive listeriosis in China, underlining the virulence potential associated with cases of invasive listeriosis (Feng et al., 2013; Zhang et al., 2016; Li et al., 2018, 2018; Fan et al., 2019). The true overall incidence of invasive listeriosis in human populations in China is, however, not known.
As only diarrhea is classified as a notifiable clinical component in China, cases of invasive listeriosis are not routinely reported to the public health authorities (Feng et al., 2010). Detection of L. monocytogenes has been reported from variety of food, such as raw and ready-to-eat meat product, milk, vegetable, yoghurt, ice-cream, and aquatic foods, suggesting a potential public health risk of infection (Yan et al., 2012; Wang et al., 2015a; Yin et al., 2015; Wu et al., 2016; Luo et al., 2017). Also, previous reports have documented the occurrence of sporadic clinical cases of L. monocytogenes in China (Feng et al., 2013; Cao et al., 2015; Wang et al., 2015b; Yin et al., 2015; Paudyal et al., 2018; Zhang et al., 2019). The origin of contamination during illness is often not known and hampers the implementation of attendant regulatory food safety and public health activities.
Multilocus sequence typing (MLST) has indicated the presence of distinct and common subtypes in human and food samples worldwide (Maury et al., 2016). However, due to the high level of genetic heterogeneity within this species, isolates can range from being putatively avirulent due to mutations or the lack of well-characterized virulence genes to highly virulent, because of the presence of lineage-specific pathogenic islands (Moura et al., 2016). Analysis of whole-genome sequences provides high resolution to confidently identify and compare bacteria isolated from different sources and information on their evolutionary relationships (Analysis et al., 2017). They also enable establishment of the context of the detected isolates to international lineages and clones.
Listeriosis is not classified as a notifiable disease in China, and thus overall information on its presence is scarce and incomplete. As a result of the relatively long incubation period (∼70 d) before the onset of severe disease and its early nonspecific clinical manifestations, the diagnosis and detection of listeriosis and its causative agent L. monocytogenes remain challenging. This makes it difficult to obtain clinical isolates on a large scale (Feng et al., 2013; Fan et al., 2019). Nevertheless contamination of retail food products sold in China with Listeria spp. is relatively high, indicating a potential risk of disease for human populations (Wu et al., 2016).
In this study, we collected and characterized 30 L. monocytogenes clinical isolates from different time points and geographical locations of China between 2002 and 2015. Whole-genome sequencing (WGS) analysis of these isolates revealed that invasive clinical disease was due to a limited number of L. monocytogenes clones and that these were highly related to the isolates that prevail in food products.
Materials and Methods
Isolates
A total of 30 isolates collected from human listeriosis cases between 2002 and 2015 of various regions in China were studied (Table 1). Whole-genome sequences for other available (as of January 2018) 13 clinical and 19 food isolates from China were obtained from NCBI and included for the analysis for this study (Cao et al., 2015; Su et al., 2017). To determine the phylogenomic context with respect to worldwide isolates of same MLST type, we downloaded all the currently (January 2018) available 10,244 genomes of all the L. monocytogenes from NCBI database. The multilocus sequence types were determined by
Details of the Listeria monocytogenes Clinical and Other Isolates Considered for the Study
Beside mentioned LIPIs, all the strains were encoding genes such as inlB, inlC, inlJ, actA, bsh, agrA, agrC, srtA, srtB, virR, virS, and fbpA that are associated with virulence.
The inlA gene was truncated in four clinical isolates in different MLST types (ST 121, 8, 87, and 9). Complete gene clusters of flagella-based motility and for chemotaxis was present in all the isolates. Antibiotic resistance gene profiling revealed the presence of several efflux pumps (lmrB, norB, lmrD, taeA, tet(42), arlR, lmrP, msrA, efrA, mdtG, lmrC, and mepA) involved in antimicrobial resistance.
A tetracycline resistance gene (tetM) was detected in one clinical isolate.
Whole-genome sequencing
For WGS, the bacterial DNA was isolated from 18 h-old cultures using MiniBEST Bacteria Genomic DNA Extraction Kit Ver.3.0 (TaKaRa) according to the instructions of the manufacturer. DNA sequencing libraries were prepared using the SPARK DNA Prep Kit (enzymatics) and VAHTSTM DNA Clean Beads (Vazyme), then paired-end sequencing with a read-length of 150 bp was performed using the Illumina NextSeq platform aiming more than 100-fold coverage.
Analysis of the whole-genome sequences
Genomic analysis was carried out by the software tool ASA3P, an in-house assembly platform (Schwengers et al., 2019). In brief, the pipeline filters reads by Trimmomatic 0.36 (Bolger et al., 2014) and uses SPAdes 3.10.1 for assembly (Bankevich et al., 2012). Contigs >500 bp were aligned to the chromosomal sequence of L. monocytogenes EGD-e and considered for further analysis. Isolates were confirmed for taxonomic allocation as L. monocytogenes by determining >95% average nucleotide identity to the L. monocytogenes EGD-e type strain (Richter et al., 2009). The average nucleotide identity of the genome was calculated by pyANI (Pritchard et al., 2016) with blastall option. Genomes were annotated by PROKAA 1.11 (Seemann, 2014), core genome was determined by Roary (Page et al., 2015), and the concatenated sequences of the core genes were used to construct phylogeny in RAxML (Stamatakis et al., 2014) with 100 bootstraps. MLST was carried out by MLST script (
Results and Discussion
Genome-based characteristics of the isolates
The draft genomes of the 30 clinical isolates varied from 2.8 to 3.1 Mb and encoded for between 2847 and 3188 genes, which is a range typical for L. monocytogenes (Supplementary Table S1). Isolates were confirmed as L. monocytogenes sensu stricto following WGS-based average nucleotide analysis (Supplementary Table S2). We extracted sequence data from public databases for an additional 13 genomes of clinical L. monocytogenes isolates from China to obtain a dataset comprising genomes from 43 isolates. All isolates harbored an intact Listeria pathogenicity island I (LIPI-I), while many isolates harbored the newly discovered lineage-specific LIPI-IV associated with neurovirulence, as well as other previously known virulence genes. Six lineage I isolates carried in addition the LIPI-III pathogenicity island harboring the Listeriolysin-S gene cluster, that encodes for bacteriocin for inhibition of competitive microflora during gut colonization (Quereda et al., 2017) (Table 1). Antibiotic resistance gene profiling revealed the presence of several efflux pumps involved in mediating resistance toward antimicrobials, and specific genetic determinants for resistance to rifamycin, elfamycin, fluoroquinolone, isoniazid, triclosan, and lipopeptide antibiotic were present in all isolates (Table 1). A tetracycline resistance gene was present in one of the human-clinical isolate.
L. monocytogenes clonal complexes −87, 8, and 9 isolates in clinical cases in China
MLST of the 43 clinical isolates identified them as belonging to ST87 (n = 12) and as members of the CC87. Other isolates were ST8 (n = 5) and ST9 (n = 4) that belonged to CC8 and CC9, respectively (Fig. 1 and Table 1). An analysis of core genome-based phylogeny using a previously characterized set of 101 L. monocytogenes isolates obtained worldwide revealed that 22 and 21 isolates belonged to lineage I and lineage II, respectively (Fig. 2). The clinical isolates from China formed distinct clusters that distributed across the phylogenetic tree of the worldwide isolates.

The phylogenomic tree and metadata of Listeria monocytogenes isolates from clinical and food isolates from China. The phylogeny is based on the concatenated sequence of the core genes (n = 1728, 1.81 Mb). Isolates of CC −1, −8, −9, −87, −121, and −155 have been the most frequent contaminant of food in China. Clinical isolates of each CC/STs were genomically closely related to the isolates of respective CC/STs from food. Bootstrap values >70% are shown at the nodes.
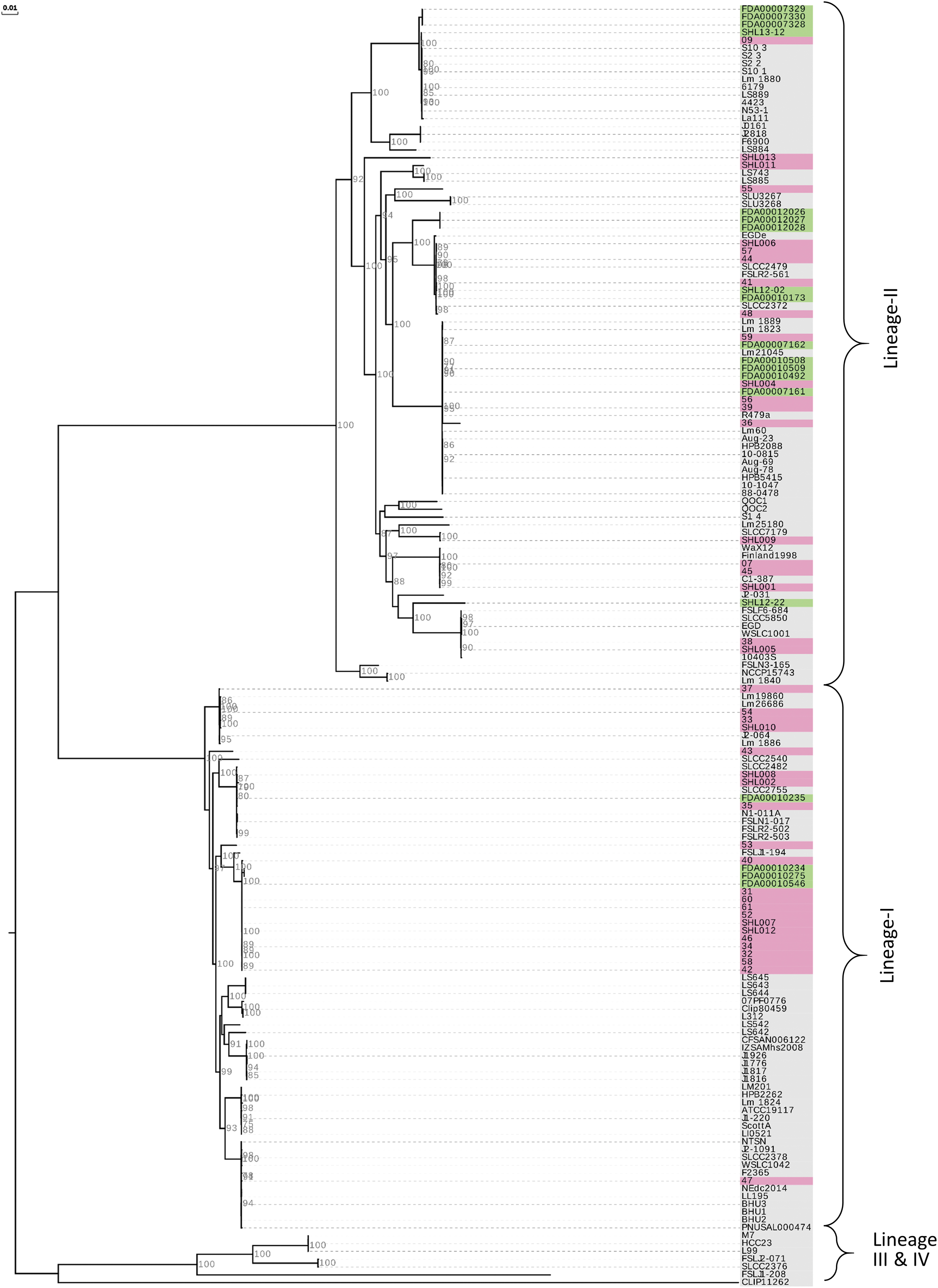
A maximum likelihood phylogenomic tree of L. monocytogenes isolates from clinical and food sources from China in comparison with isolates obtained from various sources and locations worldwide. Isolates from China (pink-clinical, green-food) formed several distinct clusters and are distributed throughout the phylogenetic tree and indicate the diverse nature of the isolates. The phylogeny was constructed using Harvest suite and is based on the core genome of ∼1.6 Mb common to all isolates. The bar represents the nucleotide substitution per site. Bootstrap values >70% are shown at the nodes.
Detailed comparative genomic analysis of CC-87, eight, and nine clinical isolates indicated that between 90% and 93% of all genes were present in isolates of a given clonal complex (CC). Moreover, nucleotide level comparisons of the core genome revealed that isolates of CC-8 differed 1099–1117 SNVs, while isolates of CC87 and CC9 differed only 18–44 and 132–182 SNVs, respectively (Supplementary Table S3). Moreover, isolates of specific CC shared the majority of the accessory genome (detailed later in the article). This high level of genomic similarity indicates the close relatedness between clinical isolates, in particular, those of CC-87 and 9.
Circulation of common clones in clinical and food sources
The three most common CCs among clinical isolates (8, 9, and 87) observed in this study have been also reported to be the most frequent CCs from a variety of food sources such as meat (mainly pork, also in beef), vegetables, sauced pickle products (of pork and beef), chicken, cooked meat, and mutton in China (Wang et al., 2012; 2015a; Wu et al., 2016; Li et al., 2018). To determine if the clinical and food isolates are related, we performed comparative analysis of the sequenced genomes of isolates from both sources. A core genome-based phylogenomic comparison clustered isolates of the respective CCs from food (CC87; n = 3, CC9; n = 2, and CC8; n = 5) and clinical sources together (Fig. 1). Genomic comparison of the isolates of each CC among food and clinical isolates revealed that they share an identical core gene set. Moreover, the overall differences in the number of SNVs among food isolates were in the same range as those of clinical isolates (Supplementary Table S3). In the case of CC87 isolates, food and clinical isolates differed only by 184 SNVs indicating a strong clonal relationship.
To understand the differences in these CCs, we also examined the compositions of their accessory genomes. For some food isolates of a given CC, we detected common prophage profiles to those of clinical isolates (Supplementary Fig. S1). Plasmids were identified in 15/19 (73.6%) food isolates. Remarkably, plasmids from food isolates were also identical to those present in clinical isolates of the respective CCs (Supplementary Fig. S2). Hence, in addition to conservation within the core genome, the composition of the accessory genome comprising prophages and plasmids in individual CCs was high. The strong overlap between clones detected in clinical cases and retail food here is unusual, it suggests the circulation of clonal isolates between both ecological compartments and the potential translocation of Lm along the food chain. Source distribution studies of L. monocytogenes samples from France and North America congruently identified CC-1, 2, 4, and 6 as the most common CCs in clinical and nonclinical sources (Maury et al., 2016; Lee et al., 2018). In contrast, our data identified CC87, 9, and 8 to be relatively predominant, while CC-1, 2, 4, and 6 were scarce, in China.
Examination of the accessory gene content from these predominant CCs identified various mobile and/or transient genetic elements and indicated that prophages and plasmids were the major drivers of intra-CC genome variability. Individual CCs harbored up to eight different types of prophages, which based on their average nucleotide identity clustered into three to five groups indicating relatively high diversity (Supplementary Fig. S1). We detected a total of 10 different plasmid types in 16 of the 43 (37.1%) clinical isolates, that is, 2 of 15 in CC87, 9 of 10 in CC8, 4 of 6 in CC9, and 2 of 4 in CC3 (Table 2). Based on the previously defined repA gene classification system for L. monocytogenes plasmids (Kuenne et al., 2010), these plasmids can be broadly placed into two groups (Fig. 3). As noted earlier, plasmids are often associated with environmental isolates (Kuenne et al., 2010; Muhterem-Uyar et al., 2018), the presence of one plasmid in 55% of foods isolates from Canada and Switzerland were reported (Hingston et al., 2017). However, a variety of plasmids were detected here from the clinical isolates harboring genes encoding for heat- and heavy-metal resistance, DNA modification-, as well as toxin-antitoxin-systems. The relatively high (16/43) occurrence of plasmids in these clinical isolates is unusual and suggest a transfer of clonal strains between these compartments. These plasmids provide advantages to the cell by elevating the tolerance abilities to environmental stresses and thus increasing the chances of survival in the food-processing environment and in contaminated food (Muhterem-Uyar et al., 2018).
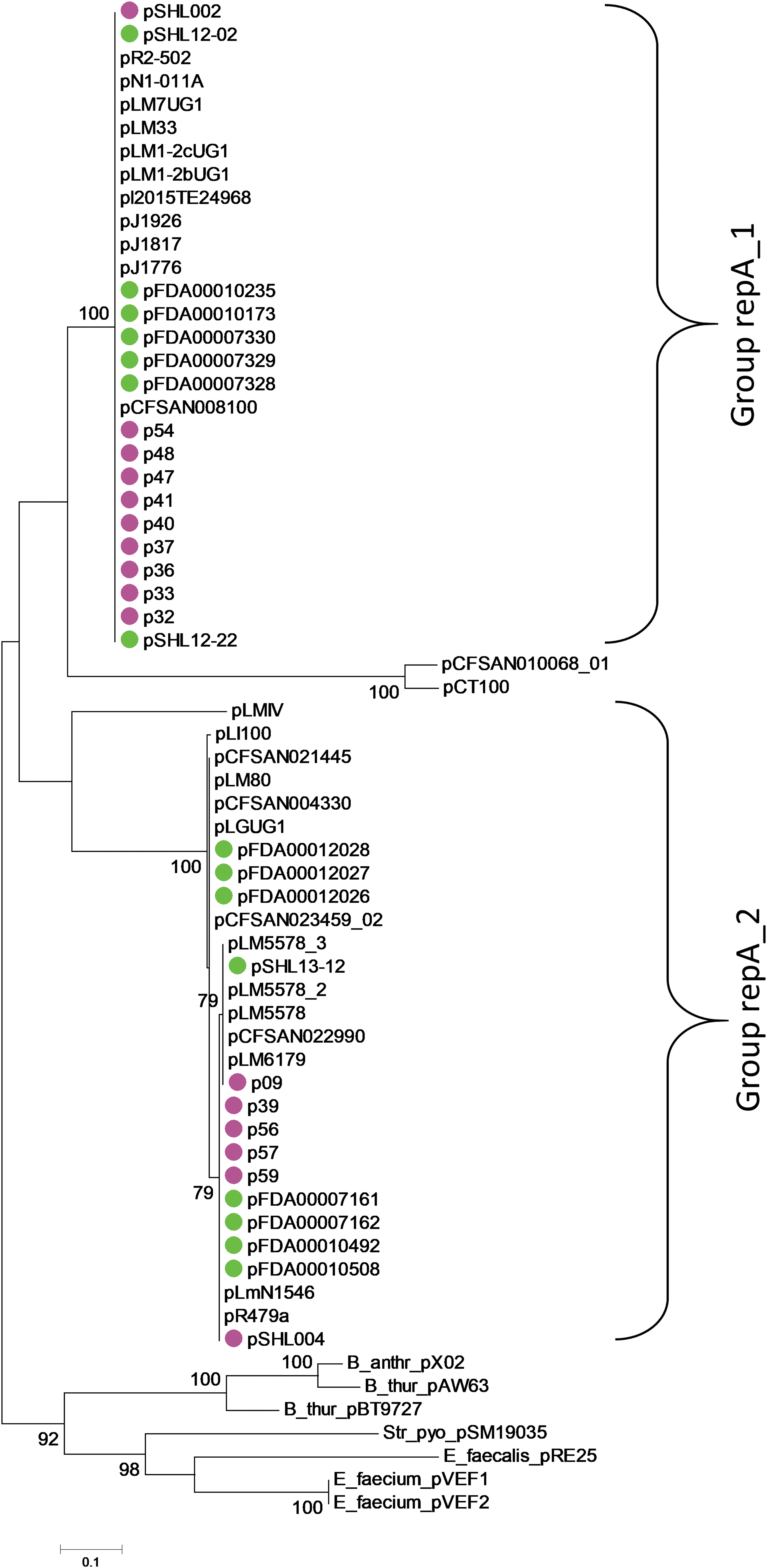
Plasmids from clinical and food isolates from China. Of the 62 isolates studied, 31 isolates (16/43 clinical and 15/19 food) harbored plasmids, which based on the repA gene sequence, showed two main groups as identified earlier (Kuenne et al., 2010). Plasmids from clinical and food isolates are denoted by pink and green filled circles, respectively. The bar represents the nucleotide substitution per site. Bootstrap values >70% are shown at the nodes.
BLAST Results for the Plasmids from Studied Isolates Against NCBI Plasmid Reference Database
The reference plasmids were identified by BLAST analysis.
The predominant L. monocytogenes ST87 clone in China is highly conserved
We studied the genomes of the predominant clone that is, ST87/CC87, in greater detail. To determine the degree of relatedness to the presently known worldwide ST87 isolates, we screened genomes of L. monocytogenes from NCBI database and extracted 82 isolates of ST87 reported from various regions of the world (Supplementary Table S4). The core gene sequence-based phylogeny grouped the majority of clinical and food isolates from China into a unified clade, while isolates from other countries grouped in different clades (Fig. 4). These data indicated that ST87 isolates from clinical and food sources in China were more closely related to one another than to the isolates from other countries. Also, clustering of Chinese ST87 isolates in a unified clade indicated their origin from a single common ancestor.
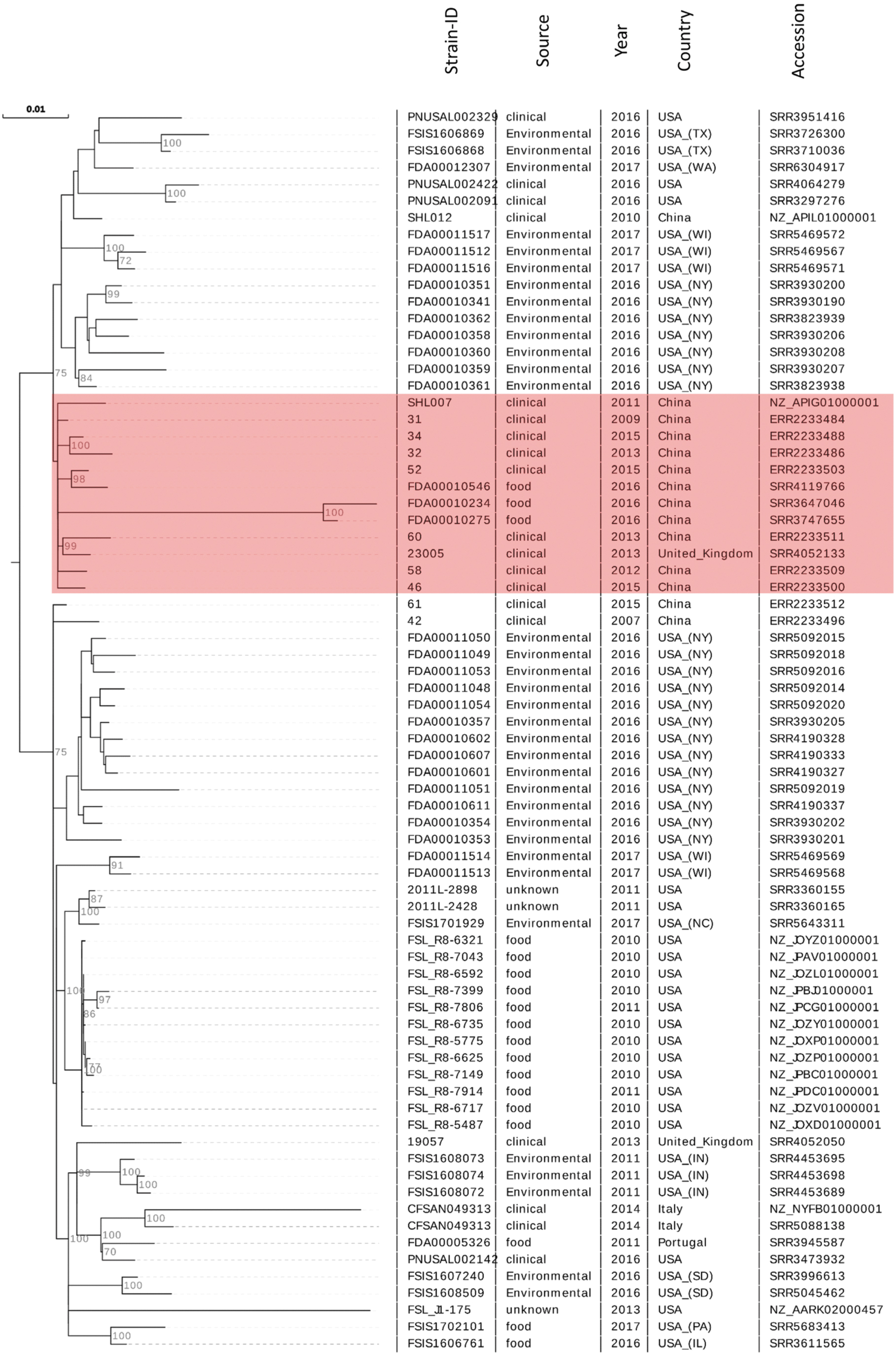
The phylogenomic tree of L. monocytogenes ST87 strains isolates from China and other countries. The tree is constructed by maximum likelihood estimation based on concatenated nucleotide sequences of the core genes (n = 2566, 2.38 Mb) using RAxML tool. Compared to the isolates from other countries, the majority (12/15) of L. monocytogenes ST87 isolates from clinical and food sources of China clustered in a single separate clade (red highlight), suggesting their close relationships.
A literature review indicated, isolates of ST87 have been reported in outbreaks from Spain and Japan (Perez-Trallero et al., 2014; Ariza-Miguel et al., 2015) with sporadic isolates reported from Colombia, Russia, Australia, Taiwan, Singapore, and USA (Huang et al., 2015; Chau et al., 2017). By comparison, the incidence of ST87 appears to be relatively high in China with the oldest clinical case dating from 2007 in Hangzhou, China. In particular, the emergence and high prevalence of L. monocytogenes ST87 isolates in both retail food and clinical cases occurred over a relatively long period of time (2002–2015). Moreover, a recent study reported high prevalence of ST87 in the feces of wild rodents (Wang et al., 2017).
The clinical isolates of ST87 in this study were obtained from cases with severe clinical manifestations such as intracranial infection, stillbirth, gastroenteritis, and septicemia. Previous reports from China record high prevalence of ST87 in food products. In this study, we demonstrate that isolates of the same ST from clinical cases and retail food are highly related both in their core and accessory genomes, and suggest transmission from food products to humans. As many ST87/CC87 isolates harbor multiple virulence determinants, including 1,3 and 4 should be in roman. LIPI-I, LIPI-III and LIPI-IV and are associated with invasive disease, increased attention is now required to both track ST87 and understand its virulence potential as well as the basis of its environmental persistence.
Conclusion
WGS was used to examine the genetic diversity of L. monocytogenes isolates obtained from invasive listeriosis cases in China. A limited number of clones of L. monocytogenes that have been circulating for an extended period in China are possibly responsible for invasive listeriosis. These clones are relatively unique to the country and are uncommon in other parts of the world. Strong identities in both the core- and accessory genome suggest circulation of clonal isolates in both food clinical disease. Taking into the account that the isolates studied were obtained over a 13-year time span and were recovered from different locations, the epidemic nature of these clones becomes apparent. Finally, our studies suggest implementation of enhanced surveillance for clinical listeriosis and implementation of effective hygienic measures in food production to prevent and control human listeriosis in China.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Key R&D Program of China (2017YFC1601201), the National Natural Science Foundation of China (No. 31472193), Key research and development program (Modern Agriculture) project of Jiangsu Province (BE2017341), the Priority academic development program of Jiangsu higher Education institutions, and by the German Federal Ministry of Education and Research (Infect-ERA PROANTILIS to T.C.).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.