
Abstract
Recently, horse meat (basashi) contaminated with Sarcocystis spp. caused food poisoning in Japan. An official detection method provided by the Ministry of Health, Labour and Welfare (MHLW), Japan, was designed to detect Sarcocystis fayeri to diagnose and control outbreaks of basashi food poisoning. In 2011, Sarcocystis-contaminated venison also caused food poisoning. However, the official MHLW detection method was not adequate for detecting Sarcocystis spp. in venison. In this study, we established a novel PCR-based detection method that amplifies 18S rRNA gene based on the conserved region of the sequence in 32 species of Sarcocystis for screening and quantification. Fifty venison samples from three areas in Hokkaido were examined by the MHLW method and the novel detection method. All samples were Sarcocystis spp.-positive. A sequence analysis indicated the presence of a species of Sarcocystis specific to sika deer (Cervus nippon), and not to horses. Another primer pair was designed for a quantitative real-time PCR assay to determine the copy number of the Sarcocystis-18S rRNA gene in parasitized venison. The melting curve analysis revealed high specificity of this assay. The calculated curve demonstrated that this quantitative PCR assay showed R 2 value of 0.993 with 10–106 copies. Using this quantitative real-time PCR assay, the gene copy numbers were determined in 50 venison samples. The copy numbers of each sample ranged from 104 to 107 per gram. The copy numbers differed according to the area in Hokkaido. This indicates that the density of Sarcocystis spp. that infect Sika deer in Hokkaido is affected by the area. The novel screening and quantitative PCR method for Sarcocystis in venison was useful for collecting epidemiological information on Sarcocystis in wild Japanese sika deer, which will contribute to improve the safety of venison products in Japan.
Introduction
More than 70
To solve these problems, local governments are taking measures to control deer populations through hunting and the promotion of the game meat industry to promote the effective use of this resource. Because wild animals are not hygienically controlled, there is a possibility of human contamination with unknown meat-borne pathogens (Kadohira et al., 2000).
The genus Sarcocystis contains protozoan species that infect herbivorous and omnivorous animals, intermediate hosts, and carnivores, the definitive hosts of these parasites. Intermediate hosts become infected by ingesting sporocysts that were excreted from definitive host in grass and water. Sporozoites excyst from sporocysts in the small intestine, then schizogony begins in the endothelium of capillaries, virtually throughout the body. Merozoites form at the periphery and then liberated from the terminal schizogony initiate sarcocyst formation. After repeated divisions, the sarcocyst is filled with bradyzoites, which is the infective stage. The whole life cycle of Sarcocystis is completed by the intake of sarcocysts in the intermediate host by the carnivorous definitive host.
Recently, several outbreaks of Sarcocystis food poisoning were reported in Japan. The first outbreak, which occurred in 2011, was caused by the consumption of Sarcocystis-contaminated raw horse meat (basashi), which is a traditional local food in Kyushu (Harada et al., 2013; Furukawa et al., 2016). After that report, the genus Sarcocystis became widely known in Japan. The second outbreak, which occurred in 2012, was caused by raw venison (Aoki et al., 2013, 2017). The vomiting and diarrhea were the major symptoms, similar in both outbreaks, but some patients also showed other clinical signs, including neurological ones (Aoki et al., 2013; Kamata et al., 2014).
The Japanese Ministry of Health, Labour and Welfare (MHLW) has provided an official method using qualitative PCR for the detection of S. fayer in horse meat to deal with outbreaks of food poisoning. However, its utility for detecting Sarcocystis spp. in venison has not been confirmed, we analyzed the whole sequence of the 18S rRNA gene of Sarcocystis in parasitized wild deer to verify the MHLW's method and design an effective detection method for cervid Sarcocystis spp. In addition, we investigated the prevalence of Sarcocystis in deer captured in three areas of Hokkaido, Japan, using this method.
Materials and Methods
Sample collection and DNA isolation from venison
Fifty venison diaphragm samples were collected from Cervus nippon yessoensis that were market hunted between March 2013 to 2014 in Hokkaido (northern area n = 15; eastern area, n = 15; southern area, n = 20). The sampling areas were located >200 km from each other. Detailed information of each sample is shown in Supplementary Table S1. All samples were collected within 2 h after hunting and delivered to cold storage at Iwate University within 48 h at 4°C. Ten grams from each venison diaphragm sample was homogenized in 30 mL of phosphate buffered saline for 1 min at 5000 rpm using Excel Auto Homogenizer (NIHONSEIKI KAISHA LTD., Tokyo, Japan). DNA was extracted from 200 μL of homogenate using a Qiagen DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) in accordance with the manufacturer's protocol. The samples used in this study were collected from legally hunted ezo deer for human consumption. All the samples, materials and method of our study does not involve human participants.
PCR using the official method to detect the 18S rRNA gene of Sarcocystis in venison
A 1100 bp DNA fragment of 18S rRNA was amplified by the MHLW's method for the detection of Sarcocystis fayeri. The method was based on conventional PCR using an EX taq Hot Start kit (Takara Bio, Tokyo, Japan) with the following primers: forward (18S1F), 5′-GGATAACCGTGGTAATTCTATG-3′; reverse (18S11R), 5′-TCCTATGTCTGGACCTGGTGAG-3′ (Pritt et al., 2008; MHLW, 2016). DNA templates were added to 20 μL reaction mixture containing 2 μL of 10 × Ex Taq Buffer, 4 μL of each dNTP (2.5 mM), 2 μL of each primer (10 mM), 0.2 μL of 5 U/μL Ex Taq HS, and 9.8 μL H2O. The following PCR conditions were applied: denaturation at 94°C for 3 min; 40 cycles of 94°C for 30 s, 60°C for 60 s, and 72°C for 60 s; and final extension at 72°C for 5 min (MHLW, 2016). The novel conventional PCR method was developed by designing the following new primer pair: forward, 5′-AGCCATGCATGTCTAAGTATAAG-3′ (Sarco18S-F); reverse, 5′-TTCCTCTAAGTGTTAAGGTTCAC-3′ (Sarco18S-R). This amplified the complete 18S rRNA (roughly 1800 bp) sequence of 32 species of Sarcocystis (Fig. 1a; Table 1). PCR was conducted using an EX taq Hot Start kit (Takara Bio) with the same components used in the amplification reaction earlier. The following cycling parameters were used: initial denaturation at 94°C for 10 min; 40 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 90 s; and final extension at 72°C for 5 min.

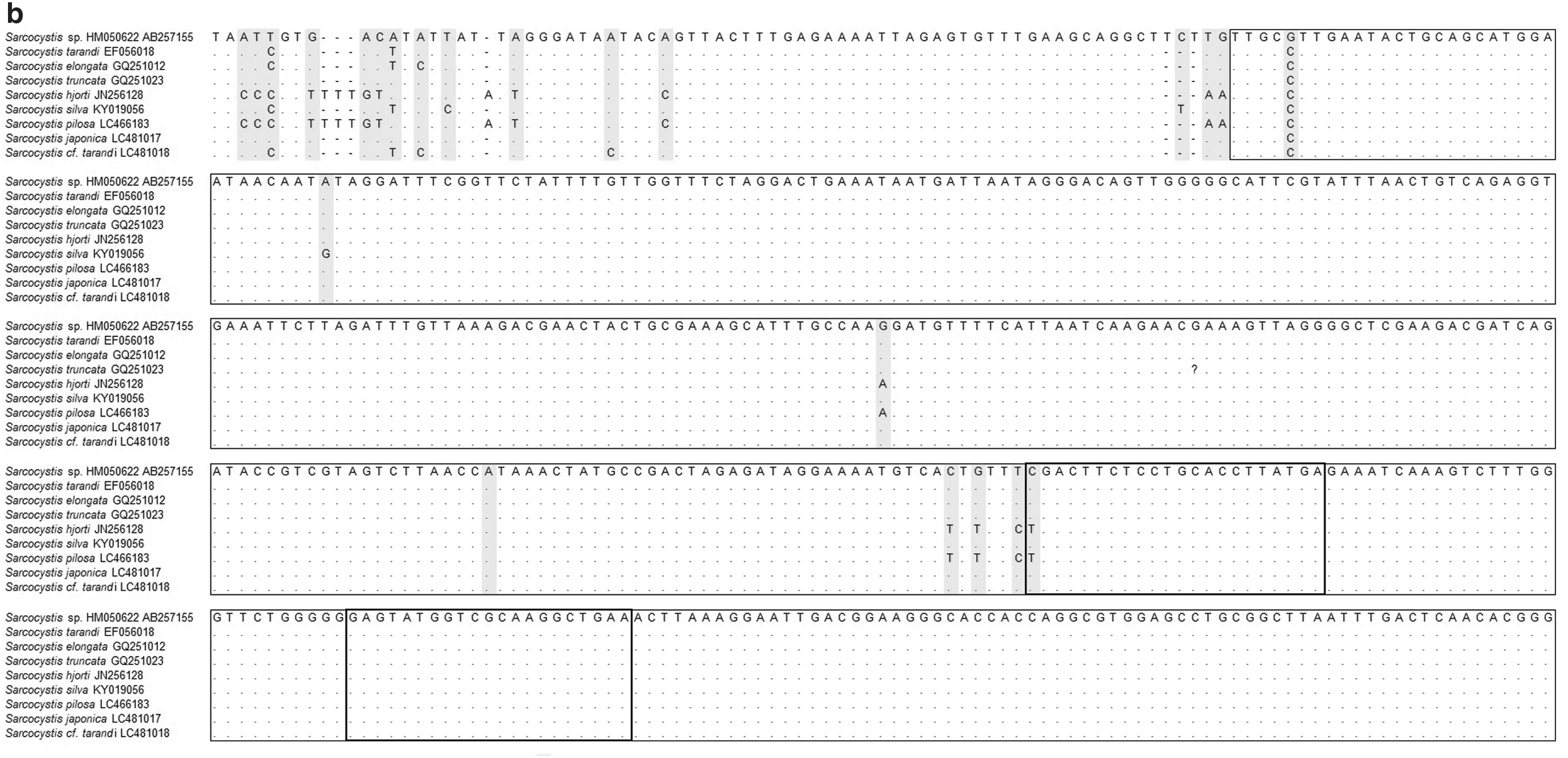
The position of the primer pairs for the amplification of the 18S rRNA gene of Sarcocystis spp.


Primers for Qualitative and Quantitative PCR Method to Amplify the 18S Ribosomal RNA Gene of Cervid Sarcocystis spp. and the Size of Amplicons Produced by Each Primer Pair
Sequencing of the 18S rRNA gene of deer Sarcocystis and quantitative PCR
A PCR amplicon was cloned into T-vector pMD20 (Takara Bio). PCR product (0.3 pmol) was added to 5 μL of reaction mixture containing 1 μL of T-Vector pMD20 and 3 μL of dH2O and then mixed thoroughly with 5 μL of DNA Ligation Kit <Mighty Mix> (Takara Bio). After incubation for 30 min at 14°C, the reaction solution added to 100 μL of Escherichia coli competent cells. The microcentrifuge tube was gently flicked for mixing and then placed on ice for 30 min. Competent cells were transformed by heat-shock for 45 s in a water bath at 42°C and then immediately returned to ice for 1 min. Super optimal broth with catabolite repression medium (S.O.C. medium) (890 μL) was added to the tube and incubated for 1 h at 37°C with shaking. Transformation culture (100 μL) was plated onto Luria-Bertani (LB)/ampicillin (final concentration: 100 μg/mL)/IPTG/X-Gal plates and then incubated overnight at 37°C. Ten white colonies were isolated and incubated in LB/ampicillin broth (final concentration: 100 μg/mL) overnight at 37°C. Plasmid vector was extracted by the innuPREP Plasmid MIDI Direct Kit (Analytik Jena, Jena, Germany). Ten clones were sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit and Applied Biosystems 3500 Series Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA). The nucleotide sequence of 18S rRNA of each clone was registered to GenBank, then applied to the basic local alignment search tool (BLAST) program of the National Center for Biotechnology Information (NCBI) (
The quantitative real-time PCR method was developed based on the common sequence of Sarcocystis spp. The 18S rRNA gene was analyzed in this study. Primer pairs for the quantitative real-time PCR assay were designed using the Primer Express 3.0 software program (Thermo Fisher Scientific) based on the common sequence of Sarcocystis spp. 18S rRNA between position 1259 to 1329 (Fig. 1b; Table 1). A SYBR qPCR kit (GeneAce SYBR qPCR Mix α; NIPPON GENE, Tokyo, Japan) was used with the following primer pair: forward, 5′-CGACTTCTCCTGCACCTTATGA-3′ (Sarcocystis Real-time F primer); reverse, 5′-TTCAGCCTTGCGACCATACTC-3′ (Sarcocystis Real-time R primer). The following PCR conditions were applied: denaturation at 95°C for 10 min, 45 cycles of 95°C for 30 s, and 60°C for 60 s. A 70 bp DNA fragment of Sarcocystis 18S rRNA was amplified from Hokkaido venison template DNA by conventional PCR using an EX taq Hot Start kit (Takara Bio) with Sarcocystis Real-time F primer and Sarcocystis Real-time R primer. The amplicon-inserted plasmid pMD20 digested by BamHI, EcoRV, EcoRV, and SalI was used as a positive control. A calculation curve was constructed using the Ct value and copy number of a positive control.
Results
Detection of Sarcocystis in venison by PCR and the analysis of the 18S rRNA gene sequence
According to the MHLW method, a sample is considered Sarcocystis-positive based on the detection of a 1100 bp amplicon. As shown in Figure 2a, various bands with various DNA sizes were observed, indicating that the PCR assay of the MHLW method would not be adequate for detecting Sarcocystis in venison. In contrast, the novel primer pair designed in this study indicated a specific amplicon of about 1800 bp (Fig. 2b).
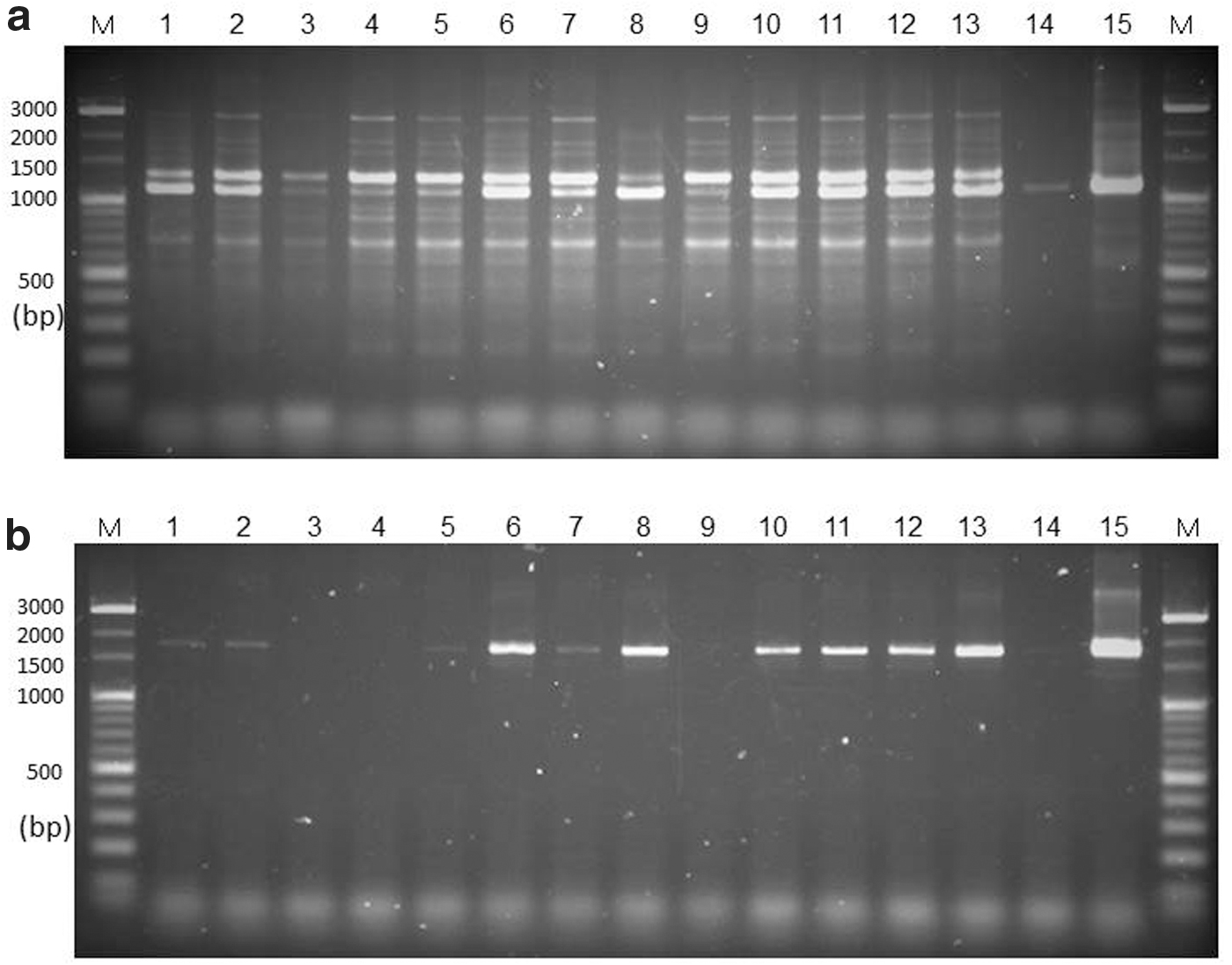
Qualitative PCR-based detection of the 18S rRNA gene of genus Sarcocystis.
Cloning and sequencing of the amplified 18S rRNA gene was performed. Six of 10 clones were successfully sequenced. The homology analysis by NCBI BLAST resulted in the identification of nine species of Sarcocystis with sequence identity >97%: S. tarandi, S. elongata, S. truncate, S. hjorti, S. silva, S. pilosa, S. cf. tarandi, S. japonica, and Sarcocystis sp. HM050622 (Supplementary Table S2) (Irie et al., 2019). In the phylogenetic analysis, each sequence of six clone is also located in a clade with the most closely related species (Supplementary Fig. S1) (Irie et al., 2019). S. fayeri was not identified from these venison samples. Among the nine species identified, a partial common sequence was found between position 968 and base 1401 in the 18S rRNA gene (Fig. 1b).
Quantitative real-time PCR to detect deer Sarcocystis and the prevalence of Sarcocystis in venison of Hokkaido
All of 50 venison samples of C. nippon yessoensis from Hokkaido were Sarcocystis spp. positive (Supplementary Table S1). A single sharp peak was observed from each of the target amplicon (70 bp) in the melting curve analysis without any other nonspecific peaks, indicating adequate specificity for the quantification of the Sarcocystis spp. 18S rRNA gene (Fig. 3a). The Ct value at each dilution and the corresponding copy numbers of the positive control indicated a correlation between 10 and 107 copies per gram with a proper coefficient of determination (R 2 = 0.993) (Fig. 3b).
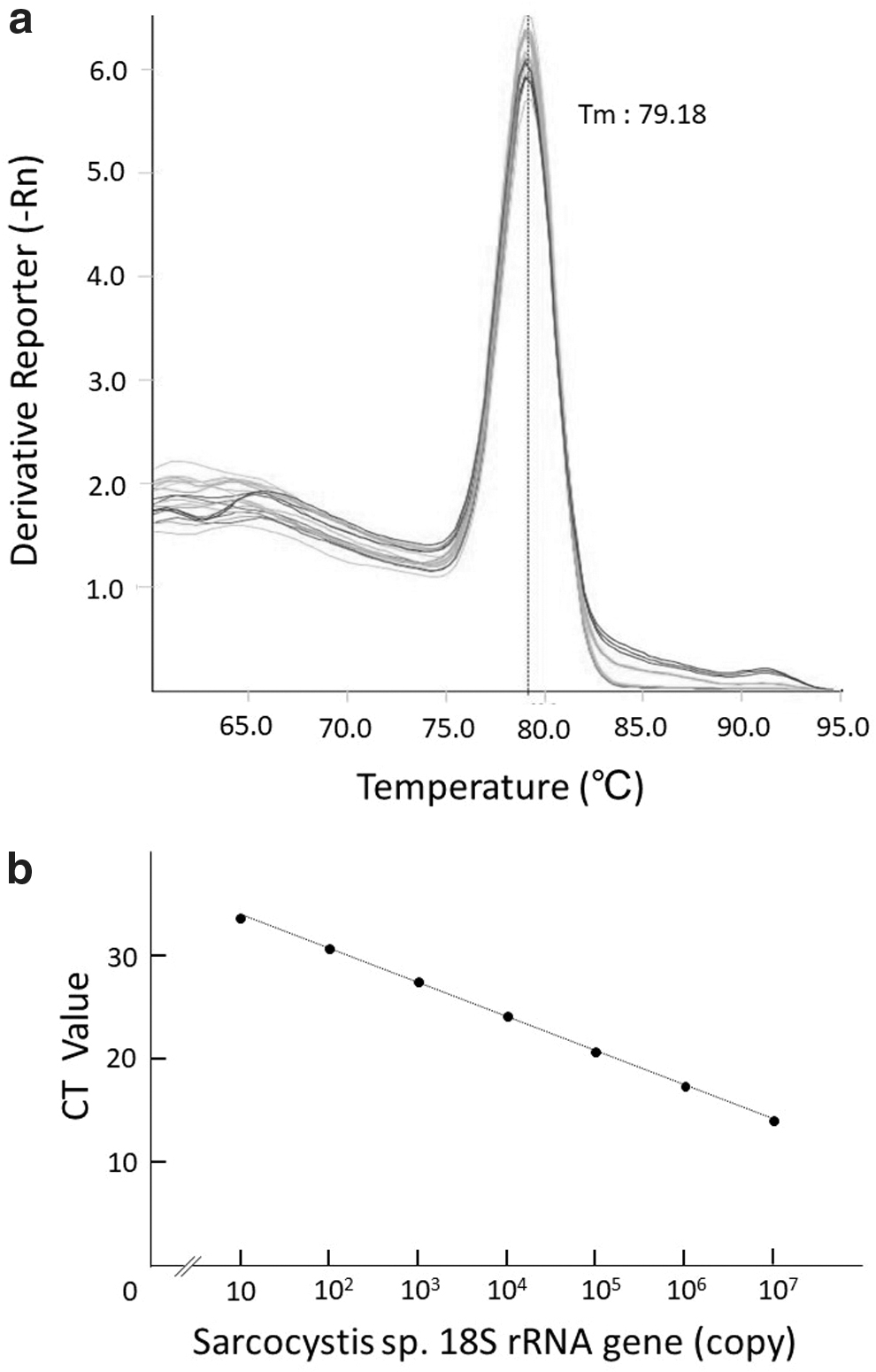
The melting curve analysis and calibration curve of the amplicon of the 18S rRNA gene of Sarcocystis spp. after the quantitative real-time PCR assay.
In total, 50 venison samples collected in Hokkaido were examined for the quantification of the copy numbers of the 18S rRNA gene of Sarcocystis spp. The median copy numbers tended to differ among the sample groups from the three sample areas in Hokkaido (Fig. 4; Supplementary Table S1). The median copy number of Sarcocystis 18S rRNA/g in venison was 105 (range 104 to 105) in the northern area. In the eastern and southern areas, the median copy numbers per gram was 106, which was 10 times the median number in the northern area; however, the range was 105 to 107 in the eastern area and 104 to >107 in the southern area (Supplementary Table S1).
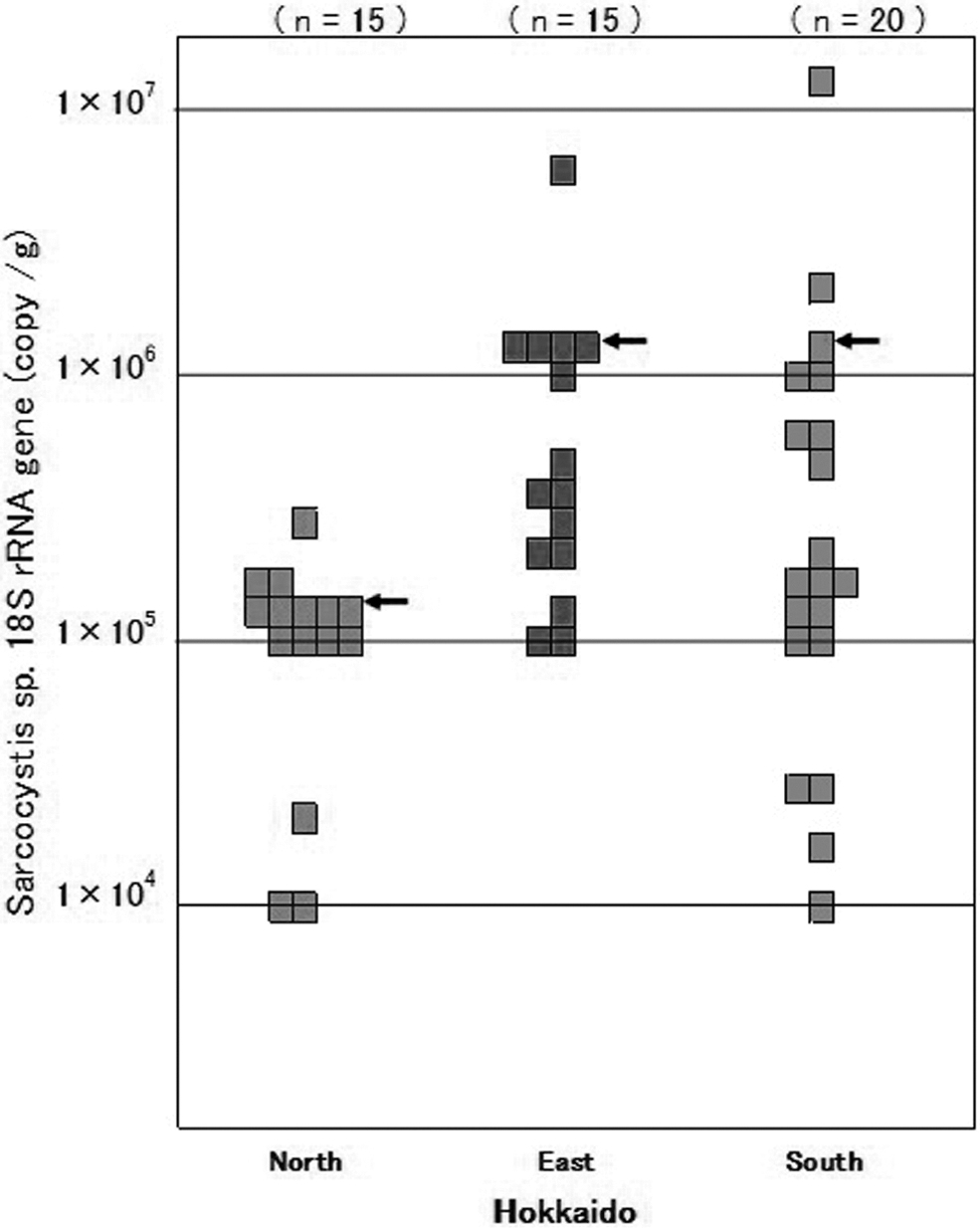
Copy numbers of the 18S rRNA gene of Sarcocystis spp. in venison samples collected in Hokkaido. The copy number of the 18S rRNA gene per one gram of sample was calculated using the quantitative real-time PCR calibration curve. The arrows indicate the median copy number of the 18S rRNA gene.
Discussion
Poor hygiene of meat is one of the main reasons for the low consumption of game in Japan. Indeed, wild species are certainly exposed to more pathogens in their natural environment than domestic livestock (Wahlstrom et al., 2003; Garcia-Sanchez et al., 2007; Jones et al., 2008; Gorski et al., 2011; Mora et al., 2012).
Inadequate information about the risk and the short history of eating game meat has led to the transmission of foodborne diseases. Some studies have reported wild cervids infected with many species of Sarcocystis (Dubey and Speer, 1985; Dahlgren and Gjerde, 2008, 2010). The MHLW official method was adapted to horse meat containing S. fayeri and its accuracy in the detection of Sarcocystis in other species of meat, such as venison, has not been assessed. To solve this problem, we tried to establish a molecular biological method to examine and quantify cervid Sarcocystis spp. in venison. First, we tried to detect Sarcocystis in venison collected in Hokkaido using the MHLW method; however, large numbers of nonspecific bands were amplified (Fig. 2a). This indicates that the MHLW method is not appropriate for other Sarcocystis spp. in venison. Thus, the new primers were designed from the alignment of 18S rRNA gene sequences of 32 species of Sarcocystis that infect herbivores (Fig. 1a). These new primers were applied to the qualitative PCR method for venison samples that were confirmed by the presence/absence of the sarcocyst under a visual inspection (Fig. 2b). As a result, the qualitative PCR using the designed primers provided an amplicon with an adequate DNA size. In addition, this method also amplified the target 18S rRNA gene from samples that were sarcocyst negative under a visual inspection (Fig. 2b). These findings indicate that the new molecular method in this study would be applicable for the detection of Sarcocystis spp. in venison.
After cloning and sequencing, nine species of Sarcocystis were identified with >97% sequence identity (Supplementary Table S2) (Irie et al., 2019). As expected, S. fayeri was not detected. The new primers were designed from these sequences of the nine species to quantify the copy number of Sarcocystis 18S rRNA gene in venison. These primers were applied to the novel quantitative real-time PCR method, clarified the distribution of Sarcocystis spp. in wild deer in three areas (north, east, and south) of Hokkaido. We found that the density of Sarcocystis tended to differ among three areas (Fig. 4).
These observations indicate that there are remarkable differences in the distribution of Sarcocystis spp. in these three areas of Hokkaido. The two cases of food poisoning due to the consumption of venison in Japan provided information about the presence of Sarcocystis in the venison for the patients; however, etiological properties, such as the species and the density of Sarcocystis parasite infection were not clarified (Aoki et al., 2013, 2017).
According to the documents provided by the MHLW and report, the causative horse meat in the case of Sarcocystis food poisoning contained 1.2 × 106 copies of the Sarcocystis fayeri 18S rRNA gene (Furukawa et al., 2016). This suggests that venison containing Sarcocystis with >106 copies per gram might cause food poisoning. In our study, we detected the 18S rRNA gene of Sarcocystis spp. within the range of 104 to 107 copies per gram in the 50 venison samples and found that 22% of the venison samples contained ≥106 copies per gram. This fact suggests that venison would harbor the same risk of food poisoning as horse meat.
In cases of S. fayeri food poisoning, diarrhea is induced by actin depolymerizing factor (ADF: a protein of 15 kDa) (Saito et al., 1995; Kamata et al., 2014; Irikura et al., 2017). Anti-S. fayeri ADF polyclonal antibody stained Sarcocystis bradyzoites in venison, meaning that Sarcocystis may also have the potential to induce food poisoning in patients consuming venison (Aoki et al., 2013, 2017). As mentioned earlier, raw venison (sikasashi) is associated with a risk of food poisoning, similarly to horse meat. Furthermore, the prevalence of Sarcocystis in venison in this study was 100%, meaning that the public risk of Sarcocystis associated with the consumption of venison would not be small. In the meantime, there is an urgent need for precise and large-scale epidemiological studies of venison, the elucidation of toxic factors, and the analysis of the mechanism of diarrhea.
The game meat industry has been promoted by the Japanese government for effective use of natural resource; however, some risk exists in relation to food poisoning from Sarcocystis and other pathogenic microbes. Other species of C. nippon are found in other areas of Japan, including C. nippon centralis in Honshu and C. nippon nippon in Kyushu (southern island of Japan). These species might harbor Sarcocystis spp., but the species and prevalence in venison would probably depend on the area. No precise information has been reported. In the future, further scientific investigations of wild deer will be needed to clarify these points.
Footnotes
Disclosure Statement
The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication. The authors declare no conflicts of interest in association with this study.
Funding Information
Our study was supported by a Health Labour Sciences Research Grant for Young Scientists (JPMHKA H30-SHOKUHIN WAKATE 003) from The Ministry of Health, Labour and Welfare, Japan.
Supplementary Material
Supplementary Figure S1
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.