
Abstract
Most outbreaks of Shiga toxin–producing Escherichia coli (STEC) are attributed to consumption of contaminated foodstuffs including beef and dairy products. In this study, we evaluated the prevalence of non-O157 STEC cultured from beef and dairy cattle and collected in Xinjiang Uygur Autonomous Region in China. Results identified 67 non-O157 STEC recovered from the 793 samples including beef cattle (10.28%, 43/418) and dairy cattle (6.40%, 24/375). A total of 67 non-O157 STEC was sequenced allowing for in silico analyses of their serotypes, virulence genes, and identification of the corresponding multilocus sequence types (STs). Twenty-one O serogroups and nine H serotypes were identified and the dominant serotype identified was O22:H8. One stx1 subtype (stx1a) and four stx2 subtypes (2a, 2b, 2c, and 2d) were found in the 67 non-O157 STEC isolates. The results revealed that stx1a+stx2a-positive STEC isolates were predominant (32.83%, 22/67), followed by stx1a+stx2d (29.85%, 20/67) and stx2a alone (17.91%, 12/67). Non-O157 STEC isolates carried virulence genes ehxA (98.51%), subA (53.73%), and cdtB (17.91%). Of the four adherence-associated genes tested, eaeA was absent, whereas lpfA and iha were present in 67 and 55 non-O157 STEC isolates, respectively. The STEC isolates were divided into 48 pulsed-field gel electrophoresis patterns and 10 STs, and ST446 (O22:H8) was the dominant clone (22.38%). Our results revealed that there was a high genetic diversity among non-O157 STEC isolated from beef and dairy cattle, some of which have potential to cause human diseases.
Introduction
Shiga toxin–producing Escherichia coli (STEC) are an important cause of foodborne disease ranging from mild nonbloody diarrhea to bloody diarrhea (BD) and hemolytic uremic syndrome (HUS). It is responsible for >1 million illnesses worldwide, resulting in nearly 13,000 disability adjusted life-years annually according to the report by FAO and WHO (FAO and WHO, 2019). According to attribution data for foodborne STEC disease burden linked to specific food categories by FAO/WHO, beef was reported to be a major food source, followed by produce (fruits and vegetables) and then dairy products (FAO and WHO, 2019). In the European Union (EU) and the United States, STEC infections were the third most commonly reported zoonosis in 2018 (Tack et al., 2019; EFSA, 2019). Compared with data from 2017, the incidents of infection increased by 39% and 26%, respectively (Tack et al., 2019; EFSA, 2019).
STEC are a phylogenetically heterogeneous large group of bacteria with >470 serotypes reported to date and ∼100 serotypes are associated with human diseases (Werber and Scheutz, 2019). Among them, STEC O157:H7 has been the most important serotype since the first outbreak caused by contaminated beef was reported in the United States in 1982 (Riley et al., 1983). However, non-O157 STEC has become an increasing cause of human intestinal disease, accounting for ∼80% of the human STEC infections recently and several serogroups such as O157, O26, O104, O45, O103, O111, O121, and O145 are commonly associated with severe illness in humans worldwide (Majowicz et al., 2014). Furthermore, some rarely reported serotypes were linked to human outbreaks including O104:H4 (Bielaszewska et al., 2011), O80:H2 (Soysal et al., 2016), and O113:H21 (Monaghan et al., 2012).
STEC are characterized by the production of Shiga toxins (Stx), an important virulence factor. There are two main types including 12 subtypes that can be further classified into three subtypes for Stx1 (Stx1a, Stx1c, and Stx1d) and nine subtypes for Stx2 (Stx2a–Stx2i) (Scheutz et al., 2012; FAO and WHO STEC EXPERT GROUP, 2019). Different toxin profiles exist among human clinical isolates of STEC and they are associated with varying probabilities of causing HUS (Werber and Scheutz, 2019). Among them, STEC producing stx2a or stx2d are most associated with HUS (FAO and WHO, 2019). In addition, other accessory virulence determinants such as intimin (eaeA) of the Locus of Enterocyte Effacement (LEE), hemolysin (hly/ehxA), and adherence (efa1/iha/lpfA) also contribute to pathogenicity (Smith et al., 2014).
Humans become infected with STEC after the ingestion of contaminated water and food, or by direct or indirect contact with human or animal feces. Overall, it has been estimated that the percentage of foodborne-acquired illnesses is 68% and 82% for STEC O157 and non-O157 STEC, respectively (Scallan et al., 2011). In the EU/European Economic Area (EEA) human infection STEC report from 2016 to 2018, the number of non-O157 STEC infections continued to increase, from 60% to 65% (EFSA, 2019). According to FoodNet in the United States, between 2010 and 2014, 5234 STEC infections were screened, of which 55% were non-O157 STEC cases (Hadler et al., 2018). After the outbreak of non-O157 STEC (O104) in Germany, the milk poisoning incident that occurred in Germany in 2017 was caused by STEC O103:H2, and it was found that the patterns of infection strains pulsed-field gel electrophoresis (PFGE) and whole-genome sequencing (WGS) were consistent with the strains in cow feces (Mylius et al., 2018). In 2018, outbreak caused by romaine lettuce was contaminated with STEC O157 and non-O157 STEC (O61) in 37 states in the United States (Bottichio et al., 2019).
According to reports, a variety of animals have become the repository of STEC around the world (Kim et al., 2020). The biggest STEC O157:H7 outbreak in China occurred in 1999 and was caused by courtyard animals leading to 195 patients being hospitalized with HUS and 177 deaths (Xiong et al., 2012). Since this time, few cases were reported (Fu et al., 2017). Nonetheless, surveillance data on STEC in ruminant animals (pigs and sheep) (Meng et al., 2014) and various foods (Bai et al., 2015) are available for some regions of China.
The prevalence and characteristics of STEC from cattle have until now only been reported in Central China (Peng et al., 2019). Similar research on STEC in Xinjiang Uygur Autonomous Region (Xinjiang) in the northwestern China, the fifth and second biggest producer of beef cattle and dairy cattle in China, is limited. In this study, we isolated and characterized non-O157 STEC isolates by PFGE analysis and WGS from beef and dairy cattle in six cities in Xinjiang with WGS, China.
Materials and Methods
Sample collection
From March 2016 to March 2017, a total of 793 samples (418 rectal swab samples of beef cattle and 375 rectal swab samples of dairy cattle) from six cities in the Xinjiang Uygur Autonomous Region (Ili, Shihezi, Changji, Wujiaqu, Urumqi, and Bole) northwestern China were taken (Ding et al., 2017; Su et al., 2017). All were collected using sterile cotton swab, and then placed in a tube containing 3 mL EC broth while being maintained on ice during transport back to the laboratory for pretreatment. The detailed sample information on pasture-based farms in this study is given in Supplementary Table S1.
Bacterial culture and strain isolation
The samples were incubated overnight at 37°C. After incubation, this enrichment broth was checked for stx (stx1 and/or stx2) genotypes by polymerase chain reaction (PCR). For the positive detections, without the step of immunomagnetic separation (IMS), a 10-fold serial dilution was prepared in sterile, distilled water and 100 μL aliquots of each dilution spread-plated onto MacConkey agar. Presumptive non-O157 E. coli (pink five colored colonies) were subcultured onto MacConkey agar and reconfirmed by PCR targeting stx genes (Scheutz et al., 2012). Then stx-positive strains were further confirmed by biochemical identification. All confirmed isolates were kept in brain–heart infusion broth with 50% glycerol in a −70°C freezer for subsequent phenotypic and genotypic characterization (Supplementary Table S2). Two reference strains (E. coli EDL933 and MG1655) were also included.
Pulsed-field gel electrophoresis
Genomic DNA for PFGE was prepared in accordance with the method described previously (Ribot et al., 2006). The PFGE patterns were interpreted with Bionumerics software (Applied Math, Belgium) using unweighted-pair group method with arithmetic mean (UPGMA). Bands that were <20.5 kb were not included in the analysis. Patterns indistinguishable by computer and visual inspection were assigned the same pattern designation.
In silico serotyping, detection of virulence genes and multilocus sequence typing
Genomic DNA was extracted using the Wizard® Genomic DNA Purification Kit (Promega, Madison, WI). WGS was carried out with Illumina NovaSeq 6000 platform (Illumina, San Diego, CA), which generated 150-bp paired-end reads from a library with an average insert size of 350 bp. For each isolate, >550 Mbp high-quality clean reads were obtained and de novo assembled using SPAdes v.3.9.0 (Bankevich et al., 2012). Serotypes were determined in silico using the SerotypeFinder tool (Joensen et al., 2015). A set of virulence genes (stx, subA, astA, cdtB, sta1, stb, senB, lpfA, iha, espP, espI, epeA, pic, sigA, cba, celB, cma, mcmA, mchB, mchC, and mchF) were also identified using VirulenceFinder (Joensen et al., 2014). In silico multilocus sequence typing (MLST) and serotyping were performed using SRST2 (Inouye et al., 2014). The seven housekeeping genes used for allele profiling included adk, fumC, gyrB, icd, mdh, purA, and recA.
Phylogenetic analysis
To uncover the relationships of our isolates with those from other countries, we obtained the STEC genomes from GenBank database. The genomes of the sequence types (STs) that our isolates belonged to were all enrolled, and two genomes were randomly selected from each of other STs. In total, we enrolled 320 genomes of 197 STs. The genome sequences of our isolates and the enrolled published genomes were compared with the complete genome sequences of E. coli strain Xuzhou21 (accession number: NC_017906) to obtain the single nucleotide polymorphisms (SNPs) using MUMmer v3.1 (Marçais et al., 2018). Then the variant sites were combined together according to the reference, and the adjacent mutations within 5 bp were filtered to avoid recombination. The concatenated sequences of filtered polymorphic variant sites conserved in all strains (core genome SNPs [cgSNPs]) were used to perform phylogenetic analysis using maximum likelihood method by FastTree v2.1.10 (Price et al., 2009).
Results
Distribution of non-O157 STEC in food producing in selected areas of Xinjiang
From the 793 samples studied, 67 (8.45%, 67/793) were identified as non-O157 STEC. By breed, 43 (10.28%, 43/418) were isolated from beef cattle, whereas 24 (6.40%, 24/375) were isolated from dairy cattle. The positive rate ranged from 3.45% to 15.38% in different cities (Supplementary Table S3). The majority of STEC (64.18%, 43/67) harbored both stx1 and stx2 genes, whereas a smaller proportion of isolates possessed only stx1 or stx2, corresponding to 13.43% (9/67) and 22.39% (15/67), respectively (Table 1).
Number of Isolates for Shiga-Toxins Virulence Genes Detected by Whole-Genome Sequencing
n, number of isolates.
PFGE analysis of non-O157 STEC study collection
All 67 isolates were analyzed using PFGE, wherein genomic DNA was digested with XbaI and the resulting banding patterns differentiated into 48 PFGE pulsotypes with 1.5% optimization and tolerance, indicating a high level of diversity from different sampling regions (Fig. 1). Eleven pulsotypes demonstrated indistinguishable patterns with the number of isolates therein ranging from two to eight and many of these were from the same cities (Fig. 1). The predominant pulsotype was identified as ECOX11.CN018, and which included eight isolates from Changji, whereas four pulsotypes (denoted as ECOX11.CN019, CN022, CN025, and ECN043) contained isolates from different cities.
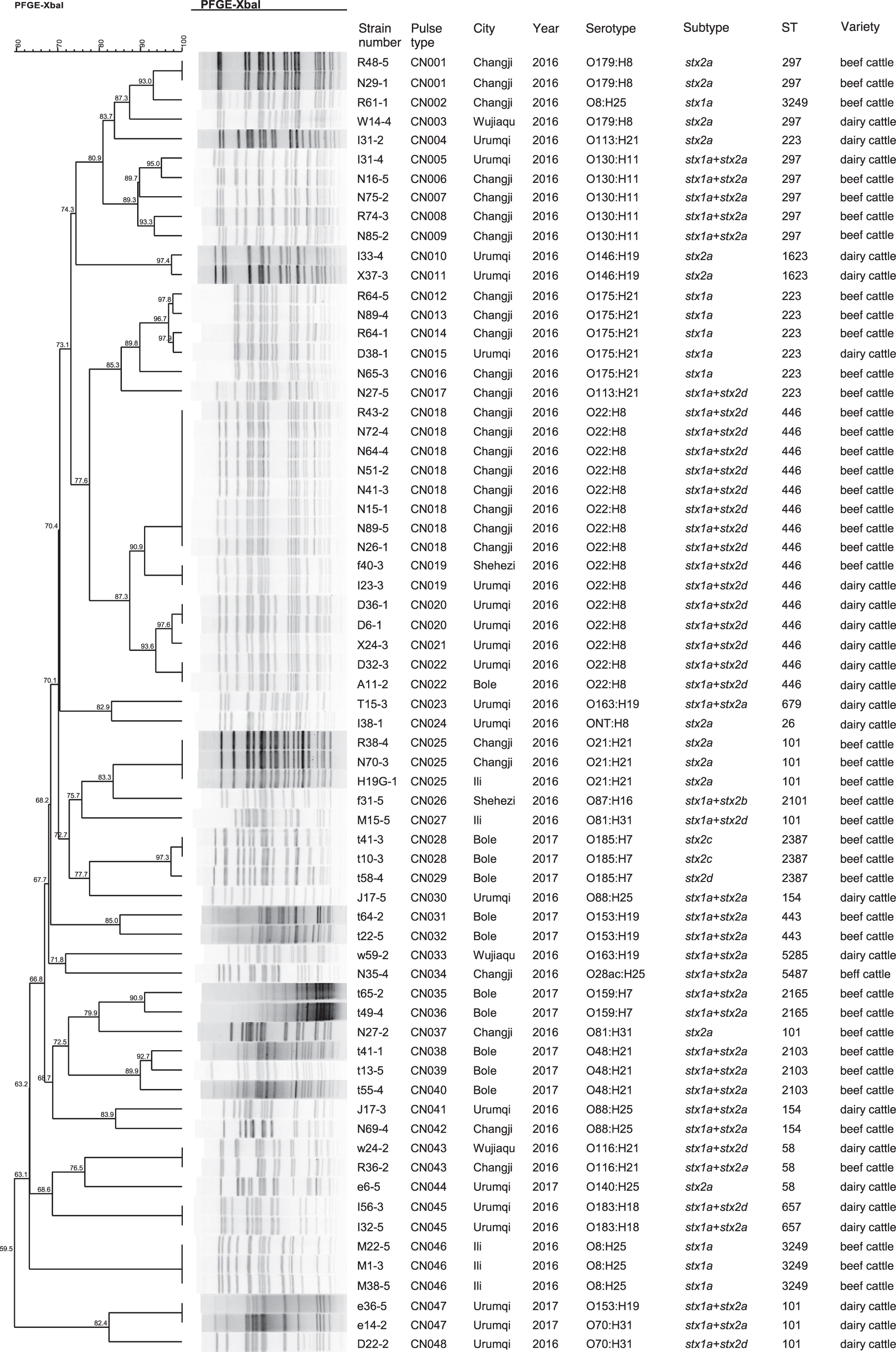
Dendrogram of 67 STEC strains isolated from cattle constructed based on PFGE with XbaI. Strains ID were indicated along with isolate pulsotype, city, year, serotype, subtype, ST and origins. PFGE, pulsed-field gel electrophoresis; ST, sequence type; STEC, Shiga toxin–producing Escherichia coli.
Serotypes, toxins, and virulence gene content of non-O157 STEC study collection
A total of 22 different serotypes along with one (E. coli I38-1) untyped were identified among the 67 non-O157 STEC isolates, including 21 different O serogroups and 9 different H types. The most frequent serotype identified was O22:H8 (n = 15) followed by O130:H11 (n = 5) and O175:H21 (n = 5). The remaining serotypes were represented by one and four isolates. Only seven serotypes were found in isolates from both beef and dairy cattle, such as O22:H8, O130:H11, and O175:H21.
Virulence and stx-encoding genes were identified using VirulenceFinder tool. The combination of both stx genes is the dominant toxin type recorded (64.18%), followed by single toxin-encoding genes including stx2 (22.39%) and stx1 (13.43%) (Table 1). Of the 52 stx1-positive non-O157 STEC isolates, all belonged to subtype stx1a. Four stx2 subtypes were identified from the 58 stx2-positive STEC isolates: 34 stx2a, 21 stx2d, 2 stx2c, and 1 stx2b (Table 1). Seven different combinations of stx1/stx2 subtypes were identified in the 67 non-O157 STEC isolates. The results revealed that stx1a+stx2a-positive non-O157 STEC isolates were predominant (32.83%, 22/67), followed by stx1a+stx2d (29.85%, 20/67), stx2a only (17.91%, 12/67), stx1a only (13.43%, 9/67), stx2c only (2.99%, 2/67), stx1a+stx2b (1.49%, 1/67), and stx2d only (1.49%, 1/67). Three of them (stx2c, stx2d, and stx1a+stx2b) were present only in non-O157 STEC isolates from beef cattle, whereas other subtypes existed in both.
The presence of 17 virulence genes was identified by WGS. None of the isolates harbored the eaeA gene. Among 67 LEE-negative non-O157 STEC isolates, most of the virulence genes detected were found to encode toxins. The number of virulence genes in each non-O157 STEC varied between 4 and 11. The frequencies of toxin genes identified were as follows: hemolysin A (ehxA; n = 66, 98.51%), subtilase cytotoxin (subA; n = 36, 53.73%), interaction of the holotoxin with the target cell (cdtB; n = 12, 17.91%), serine protease autotransporters of Enterobacteriaceae: espP (n = 57, 85.07%), epeA (n = 18, 26.87%), and espl (n = 2, 2.99%). The following rates were observed for microcins: mchF (n = 4, 5.97%), mchB (n = 2, 2.99%), and mchC (n = 1, 1.49%). The distribution of adhesins was as follows: lpfA (n = 67, 100%) and iha (n = 55, 82.09%). The positive rate of colicins was as follows: celB (n = 29, 43.28%) and cba (n = 4, 5.97%). The following proportions were observed for type III translocated genes: espA (n = 12, 17.91%). Genes related to the bacteria's ability to resist extreme acidity were identified and include glutamate decarboxylase (gad; n = 64, 95.52%). Other genes associated with the virulence system of E. coli have also been identified, including ireA (n = 1; 1.49%), which is involved in the metabolism of iron and iss (n = 54, 80.60%), increasing the bactericidal capacity of serum (Supplementary Table S4).
MLST and whole-genome phylogenetic analysis
A total of 18 STs were identified in 67 isolates. Among them, ST446 (n = 15), ST101 (n = 8), ST297 (n = 8), and ST223 (n = 7) were most frequently identified. The remaining 14 STs were found in 1–4 isolates. ST297 and ST101 strains showed different PFGE patterns, serotypes, and virulence gene profiles. ST446 consisted of only one serotype (O22:H8) and was identified from three different cities with five pulsotypes (identity from 87.3% to 100%). ST101 (O21:H21, O81:H31, O153:H19, and O70:H31), ST 297 (O179:H8 and O130:H11), and ST223 (O175:H21 and O113:H21) contained diverse serotypes.
Most STs differed from each other by two or more alleles, whereas three pairs of STs (ST223 and ST2103; ST58 and ST223; ST58 and ST5487) differed from each other by only one allele. Based on the 236,653 cgSNPs we obtained from the 387 genomes of 202 STs, we uncovered the phylogenic relationships of our isolates with the non-O157 STEC strains from other countries (Fig. 2). Corresponding to the results of MLST, the genomes of the same STs were clustered together, and genomes from STs differed between each other by only one allele, for example, ST223, ST2103, ST58, and ST5487. Our isolates were from four major STs, ST446, ST101, ST297, and ST223, and strains of these STs were also isolated in multiple countries. Our isolates were mainly clustered with those of the same STs isolated in North America (United States and Canada). In addition, although our isolates were all from cow and cattle, these STs have caused human infections in other countries and the isolates were phylogenetically close to each other.
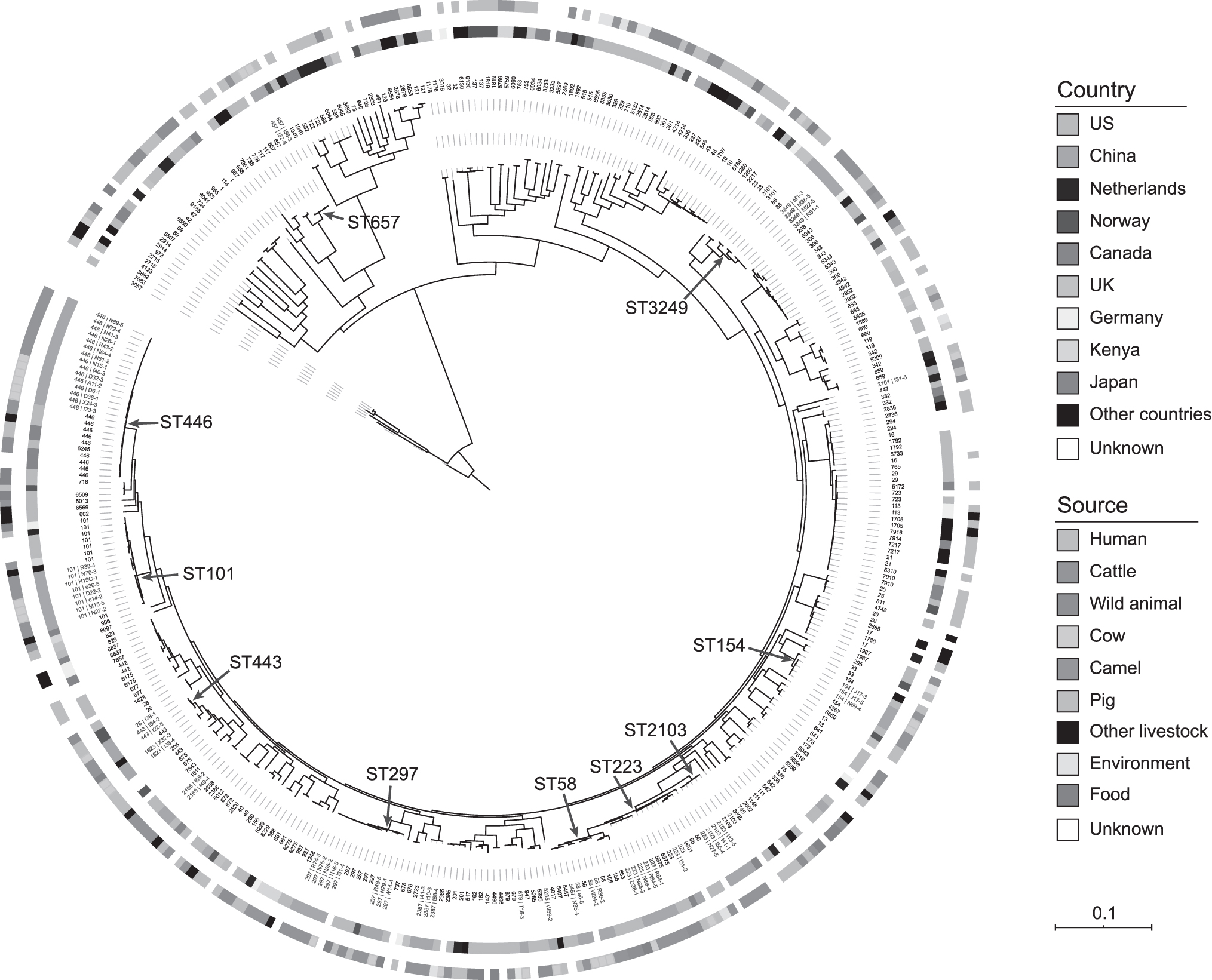
Phylogenetic tree of our isolates (n = 67) and published STEC genomes (n = 320). The STs are shown on the tips, and the isolate names of our isolates are also displayed and marked in red. The geographic origins of the genomes are showed by the ring next to the tips, and the sources are showed by the outer ring. The prevalent STs in our isolates are marked by red arrows. ST, sequence type; STEC, Shiga toxin–producing Escherichia coli.
Discussion
As the major reservoirs for non-O157 STEC, the role of cattle in these infections has been widely evaluated (Hussein and Bollinger, 2005; Hussein and Sakuma, 2005). The prevalence rates ranged widely in both beef cattle (4.6–55.9%) (Hussein, 2007) and dairy cattle (0.4–74.0%) (Hussein and Sakuma, 2005). In our study, the positive rate of non-O157 STEC from beef cattle (10.28%) or dairy cattle (6.40%) was similar with those reported in some studies (Adamu et al., 2018; Peng et al., 2019) but lower than others (Cabal et al., 2016; Hassan et al., 2017; Galarce et al., 2019). These differences might be explained by differences in sampling, sample size, methodology, geographic location, and so on (Mekata et al., 2014).
A total of 22 different serotypes along with one untypable isolate were identified in this study. Among them, most serotypes have already been described from cattle worldwide. The most common serotype found in this study was O22:H8, which has been described earlier and associated with BD and HUS (Montero et al., 2019). STEC O22:H8 isolated from cattle may act to reduce E. coli O157:H7 adherence in vitro and in vivo (Martorelli et al., 2017). Furthermore, 10 more serotypes including O21:H21 (Franzolin et al., 2005), O22:H8 (Boerlin et al., 1999), O28ac:H25 (Fratamico et al., 2010), O48:H21 (Paton et al., 2001), O113:H21(Paton et al., 2001), O159:H7 (Li et al., 2017), O153:H19 (Yamazaki et al., 1997), O163:H19 (Chart et al., 1991), O179:H8 (Beutin and Strauch, 2007), and ONT:H8 (Alonso et al., 2017) have also been previously isolated from patients with diarrhea or HUS. Of interest, none of them were assigned to the highly pathogenic serogroups (O26, O104, O45, O103, O111, O121, and O145) (FAO and WHO, 2019). One possible explanation might be that the step of IMS was not used in this study. Previous studies reported that the serogroups cannot be used as a reliable predictor of clinical outcome and all STEC strains should be considered as being pathogenic in humans, and capable of causing at least diarrhea (EFSA, 2020).
It is well known that the Shiga toxin is an important and common virulence factor identified in STEC. It is worthy of note that stx2 is more often associated with severe disease (Boerlin et al., 1999). In this study, the detection rate of stx2 (86.57%) was higher than that of stx1 (77.61%), consistent with the studies in South Africa (Mainga et al., 2018) and different from that in Chile (Galarce et al., 2019). Of the stx2 variants, multiple studies have shown that humans and cattle usually carry stx2a/2c/2d (Jost et al., 2018; Jinnerot et al., 2020), whereas stx2a and stx2d variants are more frequently associated with human disease (Orth et al., 2007; Scheutz, 2014).
Although the stx gene disrupts the function of the host's normal cells and causes cell damage, the production of stx toxin alone cannot cause serious human diseases without the adhesion of STEC strains to intestinal epithelial cells. The pathogenicity island (PAI)LEE is a major determinant of intestinal epithelium attachment of a group of STEC strains. According to 343 STEC patients surveyed in Denmark, the combined presence of eaeA and stx2 genes is an important predictor of hemolytic uremic syndrome (Ethelberg et al., 2004). However, none of the isolates harbored the gene eaeA in this study. The incidence of LEE-negative STEC strains has been increasingly reported in several countries with cumulative acquisition of PAIs (Montero et al., 2019). There may be other adhesion means and mechanisms. Putative adhesion factors include lpfA, saa, ompA, iha, and so on (Kaper et al., 2004; Ross et al., 2015), which is high in this study.
Furthermore, most of the strains (98.51%, 66/67) harbored the ehxA gene involved in the destruction of red blood cells and can cause bleeding disorders in humans (Mainga et al., 2018). The high prevalence of ehxA is consistent with the studies of STEC isolates from humans, cattle, and plants (Feng et al., 2013; Ferdous et al., 2016; Jajarmi et al., 2017; Peng et al., 2019). Gad enabling E. coli to survive gastric acidity (Castanie-cornet et al., 1999), which also increases the risk of disease in STEC. The virulence factors detected in this study have also all appeared in the relevant lineages of human STEC isolates (Ferdous et al., 2016; Shridhar et al., 2018). Moreover, a number of studies have shown that cattle and human isolates shared similar toxigenic profiles (Murinda et al., 2004; Tristão et al., 2007).
STEC O22:H8 isolates were found in both beef cattle and dairy cattle. However, it is interesting that celB gene was only present in beef cattle. It has been shown that CelB expression was sufficient to cause host cell lysis (Pugsley and Schwartz, 1983). However, STEC O22:H8 isolates were also positive for stx2d, lpfA, and gad, which are an increasing indicator for severe clinical outcome in the infected patients (Afset et al., 2006; Scheutz, 2014).
MLST has been used as a tool for non-O157 STEC identification (Ferdous et al., 2016). In this study, some isolates belonging to the same ST possessed different serotypes and produced different PFGE patterns. These findings are similar to previous reports (Bai et al., 2016). Among the STs, ST446 had the highest rate of isolation. Of note, ST446 is rarely reported in STEC. A previous study detected only two ST446 from 276 STEC food strains (González-Escalona and Kase, 2019). It should be noted that all O22:H8 isolates recovered in this study were identified as ST446, which has been previously isolated from food in Germany (Hauser et al., 2013). But in other studies, non-O157 STEC isolates O22:H8 detected from cattle in North Dakota was associated with ST145 (Isiko et al., 2015) and also in ST297/906 (Bai et al., 2016). These findings suggest that non-O157 STEC isolates with the same serogroup might possess genotypic diversity. In addition, the second most common ST297 has been reported as the most dominant ST (31.87%) in a previous study in China (Peng et al., 2019), whereas with a low prevalence (6.34%) among STEC isolates from cattle in Korea (Kang et al., 2014). The prevalence of ST297 clones should receive more attention.
Based on whole-genome phylogenetic analysis, there is no clear phylogenetic clade, which is the same as the analysis of STEC phylogeny in food regulated by FDA from 2010 to 2017 (González-Escalona and Kase, 2019). It is worth noting that the ST446, ST297, and ST223 related to the main STs in this study exist in the same clade as the human STEC strains in Canada, the United States, or Norway. And the strains in the clade also include other animal or food types. This also supports the notion of STEC transmission from animals or food to people. Non-O157 STEC from cattle or cow in this study might cause human infections.
Since the biggest STEC O157:H7 outbreak caused by farmyard animals leading to 195 patients being hospitalized with HUS with 177 deaths occurred in 1999 in China (Xiong et al., 2012), few cases have been reported (Fu et al., 2017). Rather than differences in genetic characterization to develop HUS, the risk of developing HUS also relates to demographic differences in food consumption patterns and behavior through exposure to the pathogen (Tarr et al., 2005). The evidence for geographic clustering and divergence seems to apply to both different STEC serotypes and strains within serotypes. However, there is still a big gap in our knowledge as to how these features can be fully understood and it remains an important challenge.
Conclusion
Our results report on the diversity of STEC strains circulating in food-producing animals in an important region of China. The isolates studied were found to harbor several virulence determinants related to moderate/severe illness in humans, and therefore, represent a potential risk for public health. Thus, continuous surveillance of STEC is important to not only identify the serotypes but also to detect the molecular determinants of virulence.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by National Key Research and Development Program of China (Grant No. 2017YFC1601502) and the National Natural Science Foundation of China (81402685).
Supplementary Materials
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.