
Abstract
Bacterial drug resistance is a significant food safety problem and public health threat. Plasmids carrying drug resistance genes may result in the rapid spread of resistance among different bacteria, hosts, and environments; therefore, antibiotic resistance monitoring and continuing research into the mechanisms of drug resistance are urgently needed. Southern blotting with probes for antibiotic resistance genes and even next-generation sequencing have been used previously to detect plasmid-borne resistance genes, but these approaches are complex and time-consuming. The next-generation sequencing requires strict laboratory conditions and bioinformatics analysis ability. In this study, we developed a simplified and sensitive method to detect plasmid-borne antimicrobial resistance genes and plasmid replicon types. Salmonella strains carrying plasmids of three different replicon types that contained mcr-1 and two ESBL-producing genes were used to verify the new method. The plasmids harbored by the Salmonella strains were separated by S1 nuclease treatment and pulsed-field gel electrophoresis (PFGE), then recovered and used as the templates for droplet digital polymerase chain reaction (ddPCR) to identify target genes. The target genes were present in significantly higher copy numbers on the plasmids than the background noise. These results were consistent with the plasmid sequencing results. This S1-PFGE-ddPCR method was less time-consuming to perform than Southern blot and complete plasmid sequencing. Therefore, this method represents a time-saving alternative for detecting plasmid-borne genes, and is likely to be a valuable tool for detecting coexisting plasmid-borne drug resistance genes.
Introduction
Bacterial resistance is becoming an increasingly severe public health problem in human and veterinary medicine (Wang et al., 2017). The use of antibiotics in agriculture and the inappropriate use of antibiotics in clinical practice has led to the production and spread of drug-resistant bacteria (Qiao et al., 2018). Horizontal transfer of exogenous conjugative plasmids and integrative and conjugative elements that carry drug resistance genes is the main way in which bacteria develop and spread drug resistance (Johnson and Grossman, 2015). Recombination of these horizontally transferred drug resistance genes into bacterial genomes that already contain drug resistance genes can result in multidrug-resistant strains that are resistant to antibiotics with different structures (Gillings et al., 2017; Firth et al., 2018). Bacterial resistance has made it difficult to treat or even delay the course of certain diseases in patients, resulting in excess mortality (Xia et al., 2016; Zhu et al., 2017). Therefore, measures are urgently needed to control the spread of drug-resistant bacteria.
It can be challenging to monitor the transfer of plasmids carrying drug resistance genes and determine how they contribute to bacterial drug resistance. In many cases, it is important to clarify whether drug resistance genes are carried on plasmids, as well as the plasmid replicon types. Several common approaches are used to identify plasmid-borne drug resistance genes: plasmid acquisition and drug resistance of the recipient bacteria can be testified through plasmid conjugation experiments (Gillings et al., 2017); pulsed-field gel electrophoresis (PFGE) can be performed, followed by Southern blot probing for labeled resistance genes and plasmid replication-specific genes (Xu and Johnson, 1995; Zhang et al., 2019); and the plasmid sequences from resistant strains can be acquired by combining short- and long-read sequencing (Zhang et al., 2016; Lomonaco et al., 2018; Saveliev et al., 2018). However, these methods are time-consuming, difficult to perform, have low sensitivity, and can entail high costs. In addition, in plasmid transfer experiments, if the bacterium contains multiple plasmids carrying drug resistance genes, these may be transferred into the recipient bacteria at the same time, making it difficult to accurately identify the plasmid on which the drug resistance gene is located (Guibert et al., 2018).
The droplet digital polymerase chain reaction (ddPCR) is superior to real-time fluorescence in accuracy, and is more resistant to polymerase chain reaction (PCR) inhibitors. To simplify the detection of plasmid-borne drug resistance genes, we combined PFGE and ddPCR to quickly and accurately determine identify the drug resistance genes and plasmid replicon type-specific genes present on different plasmids.
First, plasmid DNA was separated from chromosomal DNA by S1-PFGE. Then, the plasmids were gel-purified and used as template DNA to amplify and detect the presence of drug resistance genes. The plasmids carrying drug resistance genes could be quickly identified. However, plasmid DNA fragments separated by S1-PFGE are low purity, and can contain fragments of broken chromosomes and other plasmids (Kurien and Scofield, 2002). Traditional methods cannot distinguish between target genes located on the plasmid of interest versus those located on contaminating chromosomal DNA or other plasmids due to their poor sensitivity; thus, additional qualitative and quantitative methods are needed to analyze and confirm the presence of drug resistance genes on concerned plasmid.
ddPCR is a unique method for quantifying the absolute copy number of a target gene without external criteria (Sun and Joyce, 2017; Li et al., 2018), and has a higher sensitivity than fluorescence PCR (Sun and Joyce, 2017; Link-Lenczowska et al., 2018). In this study, we explored the use of PFGE combined with ddPCR, and evaluated this method using mcr-1–harboring plasmids from Salmonella as templates. Polymyxin is regarded as the last line of defense in clinical treatment (Biswas et al., 2012). However, the transferrable polymyxin resistance gene mcr-1, which was initially discovered in Escherichia coli isolated from animals and patients (Liu et al., 2016), has since spread to other E. coli strains, as well as to Klebsiella pneumoniae, Salmonella, and other bacteria from animal, food, environmental, and even patient sources (Liu et al., 2016; Moawad et al., 2018; Wang et al., 2018a). mcr-1 and its related genes (mcr-2 to -9) have been found in many plasmids of different replicon types, the most common of which are IncHI2, IncI2, and IncX4 (Sun et al., 2018; Liu et al., 2020; Zhang et al., 2019). In addition, researchers have found bacterial isolates in many countries that carry mcr genes, mainly mcr-1 and rarely mcr-2 to -9 (Nang et al., 2019). Many mcr-1-harboring plasmids may also carry multiple drug resistance genes such as those encoding CTX-M type β-lactamase genes (Dolejska et al., 2016; Wang et al., 2017; Birgy et al., 2018; Wu et al., 2018a; Liu et al., 2020), which could promote the emergence of super bacteria with pan-drug resistance (Zhang et al., 2016). In this study, we provide a method for detecting plasmid-borne drug resistance genes and rapidly identifying the plasmids that carry these genes.
Materials and Methods
Bacterial strains
Eight Salmonella strains isolated from outpatients were selected as the test strains, and the complete sequences of the plasmids carried by these strains were obtained by the short- and long-read genome sequencing (Lu et al., 2019). Strains Sa1493 and Sa2169 carried two plasmids each, and the other strains carried one plasmid each. The plasmid replicon types were identified from the sequencing data as IncHI2, IncI2, or IncX4 (Table 1). All strains harbored only one plasmid contained the mcr-1 gene, and plasmids from five of the strains carried both mcr-1 and ESBL-producing genes (bla CTX-M-14, n = 4; bla CTX-M-55, n = 1) (Table 1).
Overview of the Strains Used in This Study
+, positive; −, not found.
Design and optimization of the primers and probes
Three drug resistance genes (mcr-1, bla CTX-M-55, and bla CTX-M-14) and three plasmid replicon sequences (IncHI2, IncI2, and IncX4) were used as the detection target genes. Primers and probes for these genes were designed using Beacon Designer V 7.0 (Table 2).
Primer and Probe Sequences Used for the Droplet Digital Polymerase Chain Reaction Assay in This Study
Real-time fluorescence PCR was used to optimize the probe concentration for each reaction system. The real-time fluorescence PCR reaction mix contained: 12.5 μL GoTaq Probe qPCR Master Mix (2 × ); 1 μL (10 μmoL/L) each of the forward and reverse primers; the probe at a concentration of 50, 100, 200, 300, 400, 500, 600, or 700 nmoL/L; 1 μL template DNA; ddH20 to 20 μL. The reaction conditions were as follows: 95°C for 180 s followed by 40 cycles of 94°C for 30 s and 59°C for 40 s. FAM fluorescence values were collected in the end of the amplification.
Plasmid DNA extraction
Live bacteria were encapsulated in agarose gel, then digested with S1 nuclease (TaKaRa, Japan), after which PFGE was conducted to separate the plasmids (Bret et al., 1995). The electrophoresis conditions were as follows: pulse start time 2.16 s; pulse termination time 63.8 s; running time 19 h; electric field strength 6 V/cm. After electrophoresis, the agarose gel was stained, and then each plasmid band was purified using a large fragment recovery kit (GENECLEANR Turbo Kit) to obtain plasmid DNA template.
Droplet digital PCR
The ddPCR reaction mix contained: 10 μL ddPCR Supermix for Probes; 1.6 μL each of the forward and reverse primers (final concentration 800 nmoL/L); 0.5 μL probe (final concentration 400 nmoL/L); 1 μL template DNA (∼50–100 ng/μL), 5.3 μL ddH2O. This 20 μL reaction mix and 70 μL of droplet generator oil were loaded into the drop generator card (Bio-Rad) for each of the probes. The droplet generation reaction was conducted using a Bio-Rad QX200™ Droplet generator. Approximately 20,000 droplets were produced from each droplet formation hole. Each droplet emulsion (∼40 μL in volume) was transferred to a single well of a 96-well Eppendorf plate, which was then heated and sealed with a PX1™ PCR Plate Sealer at 180°C for 5 s. Next, PCR was performed as follows: 95°C for 10 min; 40 cycles of denaturation and annealing (94°C for 30 s; 59°C for 40 s); 98°C for 10 min. Finally, we read the droplet results using a Droplet Reader (Bio-Rad QX200 Droplet Reader). Quantitative analysis of the data was performed using QuantaSoft™ software to determine the number of positive and negative droplets per fluorophore in each sample. The Poisson distribution formula was used to calculate the number of copies of the target gene based on the number of positive and negative microdroplets. The copy numbers of plasmid type genes and genes encoding ESBL carried on the same plasmid were determined based on the number of positive droplets.
Genome and plasmid sequencing
Plasmids from the eight test strains were purified with 100 mL of liquid culture of the strain using the Qiafilter Plasmid max kit (Qiagen) as per the protocol for low-copy plasmids, and were sequenced using the HiSeq sequencer (Illumina). The plasmid sequences were determined using PacBio RSII (Pacific Biosciences) and MinION (Oxford Nanopore, Oxford, United Kingdom). For the PacBio RSII platform, a 10-kb DNA library was constructed and sequenced using single-molecule real-time sequencing technology. For the MinION platform, the library was prepared using the ONT 1D ligation sequencing kit (SQK-LSK108) with the native barcoding expansion kit (EXP-NBD103). Porechop (
Sensitivity and specificity analysis of S1-PFGE-ddPCR
The sensitivity of the method was assessed by the detection limit of ddPCR based on a series of diluted plasmid DNA of S1-PFGE, and DNA concentration were detected by Qubit (ThermoFisher, the United States). The diluted DNA was prepared by serial 10-fold dilutions to give concentrations ranging from 2.02 × 10−3 to 2.02 × 10−8 ng/μL. And 1 μL of each diluted DNA sample were added in each ddPCR and real-time fluorescence PCR reaction system. To detect the specificity in this study, the consistency of the results of S1-PFGE-ddPCR method and the plasmid sequencing method was compared.
Results
Probe concentration selection for the digital PCR assay
We used three drug-resistant genes (mcr-1, bla CTX-M-55, and bla CTX-M-14) and three plasmid replicon sequences (IncHI2, IncI2, and IncX4) to test our S1-PFEG-ddPCR strategy. The probe concentration needed to detect each of these genes was determined and optimized by real-time PCR. A probe concentration of 400 nmol/L corresponded to the highest fluorescence value and the lowest Cq value for the real-time PCR reaction. Therefore, the concentration for each probe was set at 400 nmol/L for further ddPCR assays.
Target gene identification by ddPCR
Two plasmid bands each were detected for Sa1493 and Sa2169, and one plasmid band each for the other strains, which was consistent with the plasmid sequencing results. The results of ddPCR showed that the positive and negative droplets for each of the target genes could be clearly divided into two clusters (Fig. 1A, C, E, G, I, K). The frequency distribution peaks are shown in Figure 1B, D, F, H, J, and L. The histogram showed clear separation of the negative and positive peaks for each of the test genes, and there were no apparent interference peaks, indicating that ddPCR can be used to detect antimicrobial resistance genes and identify plasmid replicon types from gel-purified DNA. Compared with S1-PFGE-Southern blot and next-generation sequencing, S1-PFGE-ddPCR method takes less time (Fig. 2).

ddPCR assay for target gene detection. Blue droplets were positive for the target gene, whereas gray droplets were negative for the target gene. Higher peaks were positive for the target gene, whereas lower peaks were negative for the target gene.
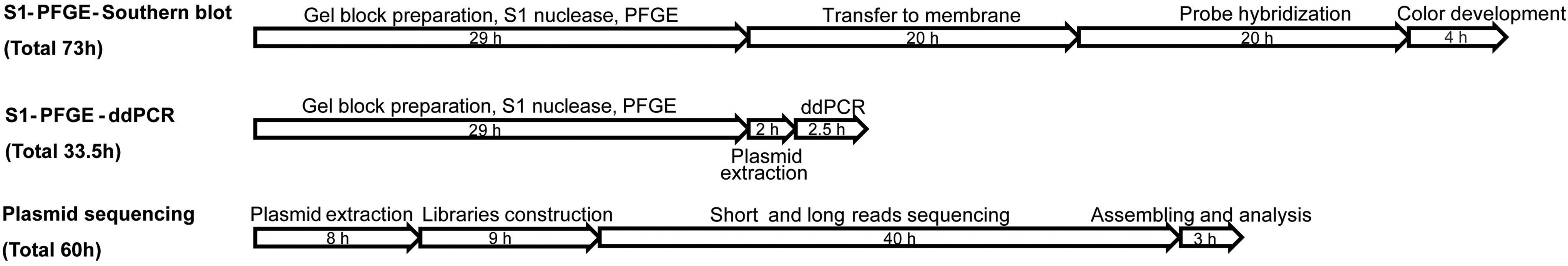
Time required to perform each method. PFGE, pulsed-field gel electrophoresis.
Determination of the plasmid replicon types
For each plasmid extracted from the PFGE gel, the replicon type genes were detected by ddPCR. The six test strains contained only one plasmid each. For each of these strains, one replicon gene was present in significantly higher copy numbers than the other genes tested (Table 3). These results were consistent with the replicon type identified based on plasmid sequencing analysis, which showed that ddPCR accurately identified the replicon type of each plasmid. Strains Sa1493 and Sa2169 both contained two plasmids (pSa1493-1 and pSa1493-2, and pSa2169-1 and pSa2169-2, respectively). The ddPCR results showed that the replicon type for pSa1493-2 was IncI2, and the replicon type for pSa2169-2 was IncX4 (Table 3), which was consistent with the plasmid sequencing results. None of the three replicon type genes tested were identified for plasmids pSa1493-1 and pSa2169-1 (Table 3). Based on the plasmid sequencing data, pSa1493-1 was replicon type IncA, and pSa2169-1 was replicon type IncCI1, which were not included as target genes in this study.
Copy Numbers of the Six Target Genes Detected in Plasmid DNA Extracted from Pulsed-Field Gel Electrophoresis Bands
The copy number of the target genes detected in plasmid DNA extracted from S1-PFGE bands was calculated as being 20 times the value obtained from the ddPCR assay, as 1 μL of DNA template was added to each 20-μL ddPCR reaction.
ddPCR, droplet digital polymerase chain reaction; PFGE, pulsed-field gel electrophoresis.
Detection of drug resistance genes
ddPCR for three drug resistance genes were performed using plasmid DNA prepared from the PFGE gel slices as template DNA. High copy numbers of mcr-1 were detected in each of the strains carrying one plasmid each, and bla CTX-M-55 and bla CTX-M-14 were also detected in some of these plasmids, demonstrating the coexistence of the ESBL-encoding and mcr-1 genes on the same plasmid (Table 3). These results were all consistent with the data obtained from the complete plasmid sequences. Strains Sa1493 and Sa2169 contained two plasmids in each. mcr-1 was detected on both pSa1493-2 and pSa2169-2, whereas ESBL-encoding genes were not detected in these two strains (Table 3). These results were consistent with the plasmid sequence data (Lu et al., 2019). We found that target genes that were actually present on the plasmids extracted from PFGE gel slices were detected in significantly higher copy numbers than background noise.
Sensitivity and specificity of S1-PFGE-ddPCR assay
According to the minimum positive copy number, the detection limit of ddPCR was 2.02 × 10−6 ng/μL (Fig. 3). The detection limit of the real-time fluorescence PCR was 2.02 × 10−4 ng/μL corresponding to 34.6Cq. Therefore, ddPCR was 100 times sensitive than real-time fluorescence PCR.
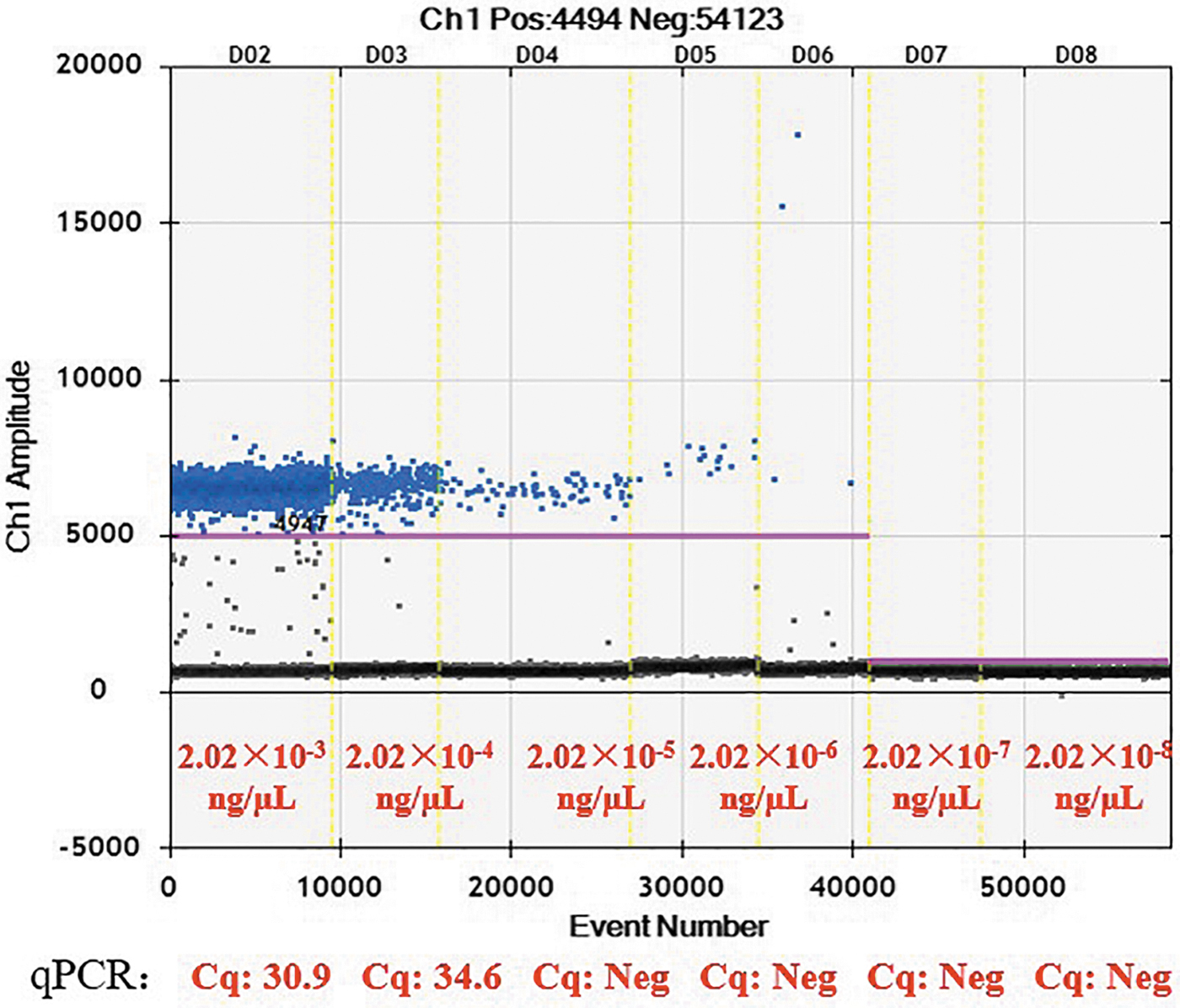
A series of diluted plasmid DNA was amplified by ddPCR. The DNA concentrations and the Cq value of real-time fluorescence PCR was shown.
The complete plasmid sequences were used as the standard, the results of S1-PFGE-ddPCR method were consistent with the complete plasmid sequences, which showed that the specificity of S1-PFGE-ddPCR method is 100%.
Discussion
Horizontal transfer of plasmids plays an important role in the spread of multidrug-resistant Gram-negative bacteria (Hardiman et al., 2016). Identifying plasmid-borne resistance genes is necessary to understand and estimate the spread of resistance among bacteria. The mcr-1 gene was first reported in 2015 (Liu et al., 2016). The global distribution of mcr-1 is well recorded (Wang et al., 2018b). mcr-1 is mainly contained by IncI2, ncxX4, and IncHI2 plasmids (Wu et al., 2018b). Detecting plasmids that carry both ESBL-producing genes and mcr-1 gene has important practical significance for food safety. The coexistence of mcr-1 and ESBL-producing genes is very common (Shafiq et al., 2019), and poses significant public health and food safety threats. In this study, we developed an accurate ddPCR method, combined with plasmid band extraction from S1-PFGE, to determine the presence of antimicrobial resistance genes on the isolated plasmids, as well as the plasmid replicon types. Commonly used methods such as S1-PFGE followed by Southern blotting with probes specific for the resistance genes are complex and time-consuming. Plasmid sequencing is also an option, but is expensive and requires a subsequent bioinformatics analysis ability (Tong et al., 2017). In this study, we verified that ddPCR can accurately determine plasmid replicon types and detect the resistance genes that the plasmids harbor without sequencing.
S1-PFGE can be used to separate bacterial plasmid DNA from bacterial chromosomal DNA. Generally, to identify the resistance genes that plasmids separated using this method harbor, the plasmid bands are transferred to a nylon membrane and hybridized with specific probes. In this study, we developed a new protocol in which S1-PFGE and plasmid band extraction was followed by ddPCR to detect high copy numbers of target genes in the extracted plasmids.
Because of the high sensitivity of ddPCR, this technique can generate background “noise” due to primer mismatch and nonspecific amplification. In addition, during S1-PFGE and plasmid DNA extraction, the target plasmid DNA can become contaminated with fragmented chromosomal DNA or with other plasmids from the same strain, which can also create ddPCR background noise (Nang et al., 2019). In our study, we observed ddPCR background noise, even though the target genes were not present in the other isolated plasmids. However, these signals were considerably lower than the positive target gene amplification signals, which were present in significantly higher copy numbers than in the negative controls, showing good discrimination.
PFGE and subsequent gel extraction can yield low amounts of plasmid DNA. ddPCR is more sensitive than ordinary PCR and real-time PCR, and thus is more sensitive than these two methods for detecting genes in small amounts of template DNA (Hindson et al., 2011).
Using labeled drug resistance gene probes, PFGE, Southern blotting, and probe hybridization are common methods for determining whether or not plasmids carry drug resistance genes. However, these methods are time-consuming, and it can be difficult to obtain definitive results due to the low membrane transfer efficiency of large plasmids (Lock et al., 2014; Lu et al., 2019). Obtaining complete plasmid sequences is an effective means of detecting resistance genes and identifying the plasmid replicon type, but this approach requires the combination of short- and long-read sequencing, which is costly and time-consuming (Postel et al., 2018). We compared the time needed to carry out the method established in this study and two other common methods and found that, compared with Southern blotting and plasmid sequencing, ddPCR is quicker and easier. Replacing ddPCR with real-time PCR could further decrease the cost of this approach, but high-quality plasmid DNA would need to be used as template DNA to ensure a high level of successful identification.
Conclusion
We believe that the strategy of combining S1-PFGE with ddPCR established in this study can replace the S1-PFGE-Southern blot method. The S1-PFGE-ddPCR method has the advantages of high detection efficiency, flexibility, simplicity, and low cost. This method may be further extended to determine whether plasmids contain any target gene of interest. It could be applied to monitor and analyze plasmid-borne drug resistance genes and drug-resistant bacteria, and could even be useful for performing resistance gene surveillance.
Footnotes
Author Contributions
Jialiang Xu and Ning Zhang contributed to all the experiments, data analysis, and article writing; Mengyu Wang and Ling Wang contributed to the data collection and article writing; Jiaqi Li, Zhe Li, and Hongqun Zhao contributed to the PFGE experiments; Ming Luo and Zhenpeng Li contributed to the data analysis; Biao Kan contributed to the article revision; Xin Lu contributed to the study conception, design, and supervision and article revision.
Acknowledgment
We thank Emily Crow, PhD, from Liwen Bianji, Edanz Editing China (
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the Foundation for Young Scholars from China CDC (2018A103), the National Science and Technology Major Project (2018ZX10714002, 2018ZX10712001-014), the Foundation from State Laboratory of Infectious Disease Prevention and Control (2019SKLID306), and Yulin Scientific Research Project (20173042).