
Abstract
Invasive listeriosis is a rare but serious foodborne disease that causes maternal–neonatal, central nervous system, and bloodstream infections. The aim of this study was to assess the whole-genome sequencing (WGS)-based genetic diversity of clinical Listeria monocytogenes isolates over a 7-year period and prove the effect of WGS application in food vehicle investigation. A total of 360 isolates were recovered during 2013 and 2019 through the national listeriosis special surveillance program. Two hundred twenty-six isolates (62.8%) were associated with pregnancy. All isolates belonged to lineage I (214 isolates) or lineage II (146 isolates), with 4 serogroups (46.9% IIb, 39.7% IIa, 12.5% IVb, and 0.8% IIc). All isolates were in 25 clonal complexes (CCs) and 3 singletons, with CC87, CC8, and CC5 being the most common causes of human listeriosis. All clinical isolates were positive for Listeria pathogenicity island 1 (LIPI-1), LIPI-3 was present in 21.4% of isolates and LIPI-4 was detected in 29.2% of isolates. LIPI-4-positive isolates, including CC87, sequence type (ST)619, ST382, CC4, and CC2, have been shown to confer hypervirulence. Fifteen isolates harbored at least one antimicrobial encoding gene, including tet (M), mef (A), msr (D), and dfr (G). The sublineage designations were consistent with CC designations, and 215 distinct cgMLST types (CTs) were classified, the most abundant being CT58 and CT750. In summary, there is a high level of genetic diversity among the clinical isolates. WGS has strengthened listeriosis surveillance and will be implemented for other foodborne bacteria in the National Molecular Tracing Network for Foodborne Disease.
Introduction
Invasive listeriosis is a rare but serious foodborne infection, mainly caused by consumption of Listeria monocytogenes (LM)-contaminated food (Goulet et al., 2008). Listeriosis is thought to induce maternal–neonatal (MN), central nervous system, and bloodstream infections in pregnant women, neonates, the elderly, and immunocompromised individuals (de Noordhout et al., 2014).
Listeriosis special surveillance was integrated into the National Foodborne Disease Surveillance Plan in 2013 in China, and all cases were reported to the National Foodborne Disease Surveillance System. Listeriosis has been classified as a notifiable foodborne disease according to the requirements of the Foodborne Disease Surveillance and Reporting Specification issued by the National Health Commission in 2019 (NHC, 2019).
The prevalence and genomic analyses of LM in food have been extensively reported in China (Liu et al., 2020; Wang et al., 2021; Yan et al., 2016; Zhang et al., 2020). However, comprehensive studies of clinical LM isolates have seldom been reported (Luo et al., 2019; Wang et al., 2015; Zhang et al., 2021). Previous studies reported the demographic and clinical characteristics of listeriosis cases (Li et al., 2019) and the role of the National Molecular Tracing Network for Foodborne Disease (TraNet) in China (Li et al., 2021; Li et al., 2018).
In this study, whole-genome sequencing (WGS) was applied to decipher the genetic diversity of clinical LM isolates over a 7-year period in 11 provinces of China and identify gene content differences between hypervirulent and hypovirulent isolates. Another aim was to verify whether WGS-derived cluster analysis can be used in foodborne disease surveillance and investigation.
Materials and Methods
Human LM isolates
From 2013 to 2019, a total of 360 human LM isolates from 97 sentinel hospitals were characterized in the framework of the listeriosis special surveillance in China. Two hundred twenty-six isolates were cultured from MN infection cases (including pregnant women, fetuses, and infants ≤28 days old), and 134 isolates were cultured from nonperinatal cases (including elderly individuals, children, and persons with chronic underlying conditions).
Only one isolate per MN case was selected for this study. Additional bacterial isolate information used in this study is listed in Supplementary Table S1. This study was approved by IRB of China National Center for Food Safety Risk Assessment. The review number is 2017016.
WGS and bioinformatic analysis
Genomic DNA extraction was performed using the DNeasy UltraClean Microbial Kit (QIAGEN), and DNA quality and quantity were assessed with NanoDrop 2000 and Qubit 2.0 instruments. Qualified DNA was dispatched to a commercial sequencing provider for library preparation and sequencing. Genomes were sequenced on the Illumina HiSeq platform using 150-bp paired-end runs with an average coverage depth of 100 times. Trimmed sequence reads were de novo assembled using SPAdes inbuilt in TraNet. Details of genome quality are presented in Supplementary Table S1.
Lineage, PCR serogroup, and sequence types (STs) were determined in silico from genome sequences using the Bacterial Isolate Genome Sequence Database (BIGSdb) platform (Jolley and Maiden, 2010). Clonal complexes (CCs) were defined into groups with single-locus variants based on multilocus sequence typing (MLST) allelic profiles.
All assembled genomes were screened for the presence of Listeria pathogenicity island 1 (LIPI-1), LIPI-3, LIPI-4, stress survival islet 1 (SSI-1), and SSI-2 using the BIGSdb-LM platform, with minimum nucleotide identity cutoff of 90% and alignment length coverage of 60% (Moura et al., 2016).
Antimicrobial resistance
Antimicrobial resistance genes and biocide and metal tolerance genes (bcrABC, cadAC, qacH[Tn6188], emrE, and qacA) were identified using the BLASTN algorithm; ResFinder database, version 3.1 (Zankari et al., 2012); and BIGSdb-LM platform with minimum nucleotide identity cutoff of 90% and alignment length coverage of 60%.
Susceptibility to a panel of 8 antibiotics was determined using the broth microdilution method, comprising penicillin, ampicillin, trimethoprim/sulfamethoxazole, meropenem, erythromycin, tetracycline, ciprofloxacin, and vancomycin (Shanghai Biofosun Biotech Co., Ltd.). Minimum inhibitory concentrations (MICs) were interpreted using the breakpoints of EUCAST, version 12.0.
Core genome MLST subtyping, core genome single nucleotide polymorphism, and phylogenetic analysis
The core genome MLST (cgMLST) scheme is based on the Institut Pasteur 1748 highly conserved core gene scheme (Moura et al., 2016). Allele calling based both on assemblies and directly on reads was performed using BioNumerics 7.6cn-based TraNet. Phylogenetic classification based on cgMLST profiles was inferred using the single-linkage algorithm. cgMLST types (CTs) were defined by the cgMLST profile that shared a maximum of 7 allelic differences of 1748 allele calls. Sublineage (SL) was determined by cgMLST profiles that differ by up to 150 allelic mismatches. The final minimum spanning tree was created using GrapeTree (Zhou et al., 2018).
The full set of 360 genomes was used to generate a core-genome single nucleotide polymorphism (cgSNP) alignment and construct a phylogenetic tree, using Parsnp, version 1.1.2 (Treangen et al., 2014). The two most dominant clades, CC87 and CC8, were selected for whole-genome single nucleotide polymorphism (wgSNP) analysis with BioNumerics 7.6. The RefSeq genomes, GCF_002043045.1 and GCF_001952775.1, were used as references for CC87 and CC8 isolates, respectively. The single nucleotide polymorphism (SNP) matrix was generated using BioNumerics with strict default filter parameters.
The wgSNP phylogenetic trees were generated with Random Axelerated Maximum Likelikhood (Stamatakis, 2014) and visualized using ggtree, version 3.2.1 (Yu, 2020).
Sequence data accession numbers
All WGS data have been deposited at the National Center for Biotechnology Information under BioProject PRJNA657119. Accession numbers for draft sequence files are included (Supplementary Table S1).
Results
Genetic diversity of human isolates
Of the 360 sequenced human isolates, 214 belonged to lineage I, 146 belonged to lineage II, and none belonged to lineages III and IV. A total of 46.9% of isolates were assigned to serogroup IIb (169/360), 39.7% to IIa (143/360), 12.5% to IVb (45/360), and 0.8% to IIc (3/360). IIb was the most common serogroup among the isolates from perinatal cases, while IIa was the most predominant among nonperinatal cases (Table 1).
Distribution of Listeria monocytogenes Clones in Clinical Source, 2013–2019 (N = 360)
CC, clonal complex; ST, sequence type.
Forty-nine different STs were identified and distributed into 25 CCs and 3 singletons. The most prevalent CCs were CC87 (78/360; 21.7%) and CC8 (44/360; 12.2%) (Fig. 1). Remarkably, CC87, CC5, and CC8 were the most abundant CCs for pregnancy-related cases, and CC87, CC8, and CC155 were the most abundant CCs for nonpregnancy-related cases (Fig. 2).
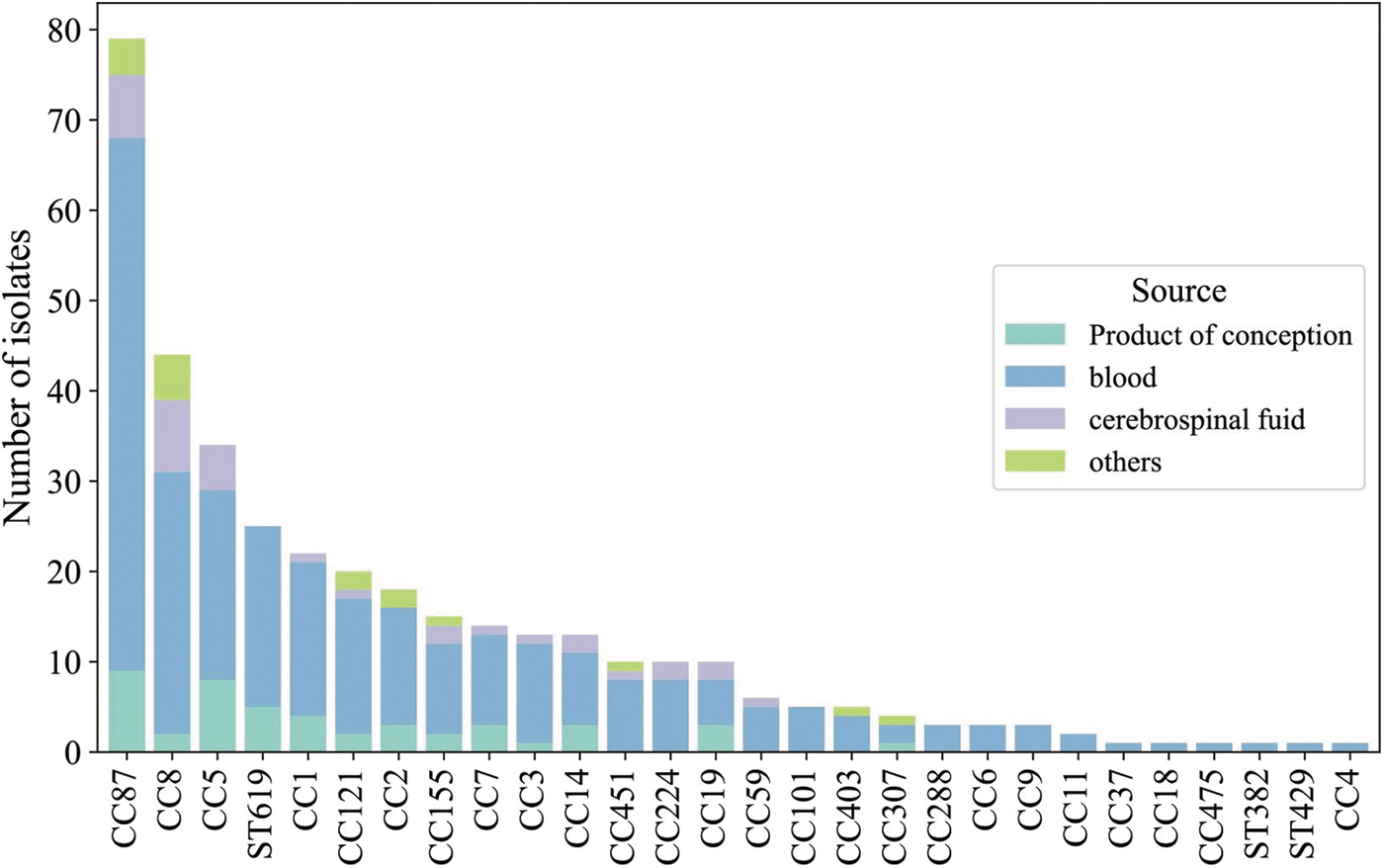
Prevalence and distribution of complex clones in different clinical sources. CC, clonal complex; ST, sequence type.
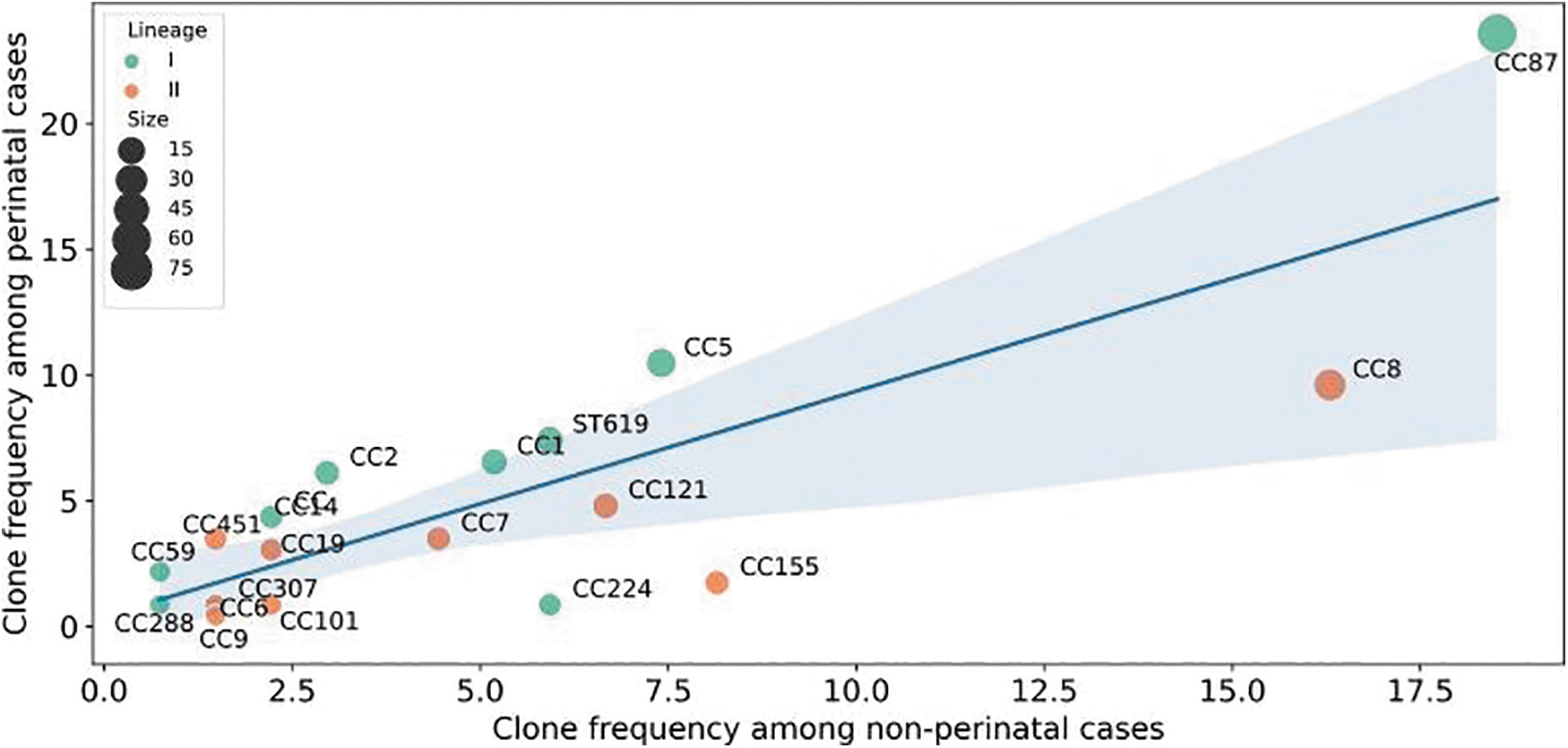
Clone frequency of isolates from perinatal and nonperinatal cases.
Certain CCs were uniquely recovered from perinatal cases (including CC403, CC11, CC475, CC382, and CC4), whereas others were exclusively isolated from nonperinatal cases (including CC429, CC37, and CC18). Nine new STs were identified and contributed to the BIGSdb-LM platform.
Antimicrobial resistance
All isolates were found to harbor the fosfomycin resistance gene, fosX. Excluding fosX, 15 (4.2%) isolates harbored at least one antimicrobial encoding gene, including dfrG, tet (M), mef (A), and msr (D). Five resistant isolates were from pregnancy-related cases and 10 isolates were from nonpregnancy-related cases. The antibiotic resistance gene profiles were concordant with results of the microdilution assay. All isolates were susceptible to the common therapeutic antibiotics, penicillin, ampicillin, and meropenem.
A total of 3 antibiotic resistance profiles were detected, including tetracycline (TET) - erythromycin (ERY), TET - trimethoprim/sulfamethoxazole (SXT), and TET, and no multidrug-resistant isolate was detected (Table 2). Among the resistant isolates, CC155 and CC87 were the most common clones; CC87 isolates were resistant to erythromycin and tetracycline harboring tet (M), mef (A), and msr (D) genes, while CC155 isolates were sensitive to erythromycin. No isolate harbored biocide resistance genes, and only 50 isolates harbored the cadA gene.
Resistance to Antibiotics in Clinical Listeria monocytogenes Isolates, 2013–2019
CTs, cgMLST types; ERY, erythromycin; MIC, minimum inhibitory concentration; SXT, trimethoprim/sulfamethoxazole; TET, tetracycline.
All 15 resistant isolates exhibited resistance to tetracycline (MICs, 16 to >32 μg/mL), carrying the ribosome protection protein gene tet (M). Seven erythromycin-resistant isolates (MICs 4 to 8 μg/mL) were detected, mainly owing to the presence of mef (A) and msr (D) genes encoding efflux pumps of the major facilitator superfamily. Three trimethoprim/sulfamethoxazole-resistant isolates were detected at high levels (MIC >8/152 μg/mL) due to acquisition of the Tn6198 plasmid-mediated dfrG gene.
Virulence factor profiles
All isolates harbored LIPI-1, the prfA-dependent virulence gene cluster. LIPI-3 was present in 21.4% (77/360) of isolates, all belonging to lineage I (Clayton et al., 2011). LIPI-3 was ubiquitous in ST619, CC1, CC3, CC224, CC288, CC6, CC4, and ST382 and present in 29.6% (50/169) of IIb isolates and 60.0% (27/45) of IVb isolates.
LIPI-4 was detected in 29.2% (105/360) of isolates, including six genes involved in carbohydrate metabolism (Maury et al., 2016). LIPI-4 appeared to be strongly associated with all CC87, ST619, ST382, and CC4 isolates within lineage I. CC87 and ST619 were the most prevalent hypervirulent human isolates in China, and ST619, CC4, and ST382 harbored both LIPI-3 and LIPI-4.
SSI-1 was present in a total of 131 isolates, which was observed in CC5, CC3, and CC224 of lineage I and CC8, CC155, CC7, CC9, CC403, CC307, and CC18 of lineage II. SSI-1 has been linked to biofilm formation (Keeney et al., 2018) and growth of LM under suboptimal conditions (Ryan et al., 2010). Only and all CC121 isolates harbored SSI-2, which is involved in survival under alkaline and oxidative stresses (Harter et al., 2017).
Phylogenetic structure and SLs
Thirty-two different SLs were designated from cgMLST profiles. Most SLs were concordant with CCs, and the dominant SLs were SL87 and SL8. Two hundred fifteen distinct CTs were classified, the most abundant being CT58 (n = 39), followed by CT750 (n = 24) and CT8370 (n = 10), which belonged to SL87, SL8, and SL619, respectively. CT58 and CT750 have been repeatedly isolated from humans over the past 5 or 6 years, suggesting their potential prevalence.
A total of 43 cgMLST-based clusters were identified, and no outbreak was confirmed through epidemiological investigations; however, 3 sporadic cases were successfully linked to specific contaminated food. One child case was caused by an ice cream popsicle; one leukemia-related case was caused by a prepackaged, ready-to-eat (RTE) meat product sold in a supermarket; and one pregnancy-related case was caused by a Chinese salad sold in a market.
A cgMLST profile-based comparison was performed to define clonal characteristics of isolates at the microevolutionary level, showing that there was a clear delineation between SLs and CCs (Fig. 3).
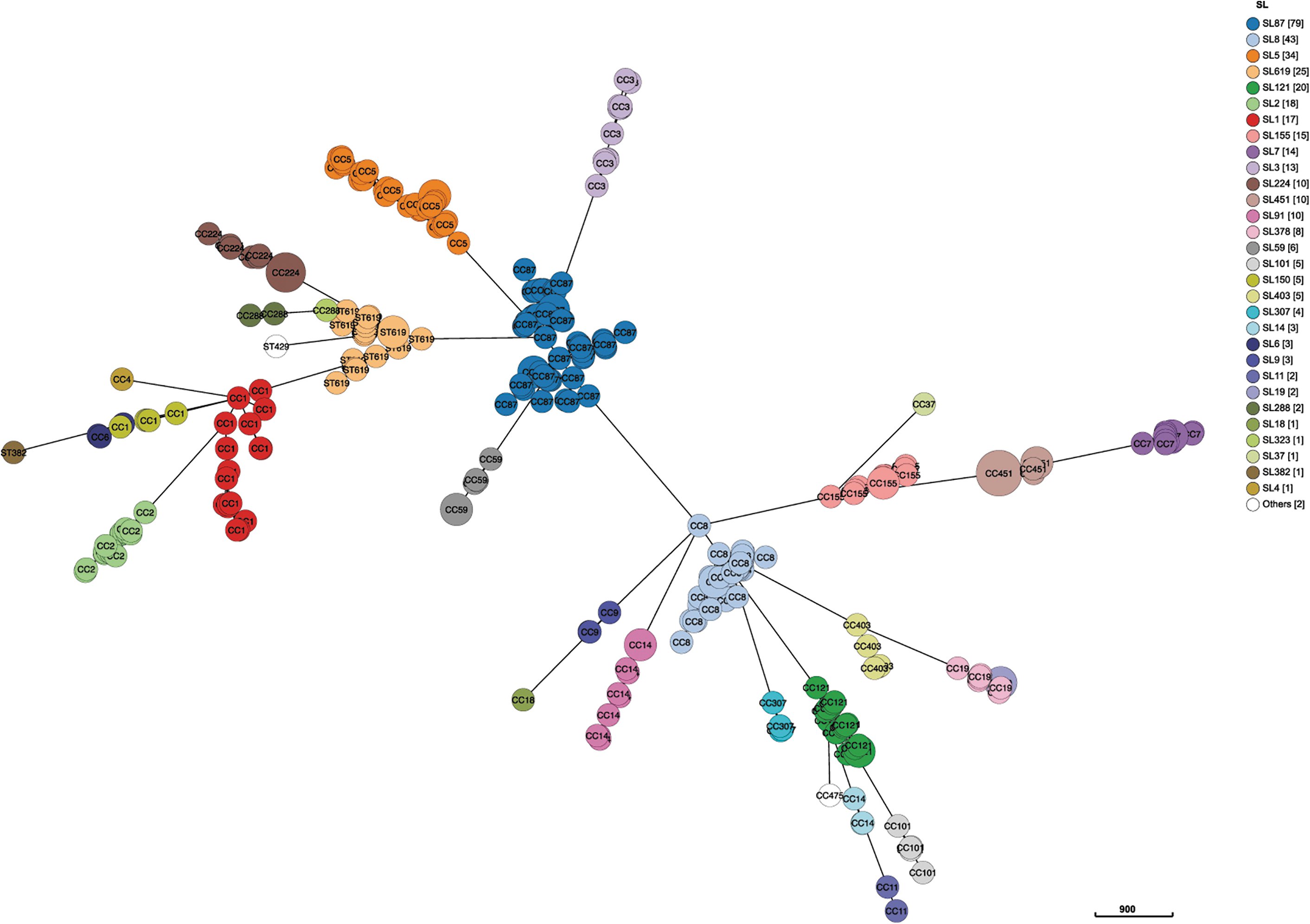
Minimum spanning tree based on clinical Listeria monocytogenes core genome multilocus sequence typing profiles. SL, sublineage.
To investigate the phylogenetic relationship, we constructed a maximum likelihood tree of the variations in core genomes of the 360 isolates. The phylogenetic tree was mainly divided into two clades that were in accordance with the lineage distribution (Fig. 4A). CC87 (n = 78) and CC8 (n = 44) clades were selected for further analysis, and CC8 isolates only harbored LIPI-1, while LIPI-1 and LIPI-4 coexisted in CC87 isolates (Fig. 4B).

Phylogenetic tree and metadata of 360 clinical Listeria monocytogenes isolates in China

Discussion
Microbial food safety has a great impact on health in China (Yue et al., 2021). An increasing proportion of the population will be at risk for LM infection due to loosening of the family planning policy in China and more women preparing for pregnancy. Moreover, new forms of prepackaged, RTE fast-food products extend the shelf life in refrigerators and increase the risk for vulnerable people. In this study, WGS was applied as a surveillance tool for deciphering molecular characteristics of clinical LM isolates in China.
Antimicrobial chemotherapy is the necessary treatment for invasive listeriosis, and LM is well known for susceptibility to a wide range of antibiotics; nonetheless, LM is intrinsically resistant to broad-spectrum cephalosporins (Morvan et al., 2010). Third-generation cephalosporins are generally used empirically in therapy, leading to delayed effective treatment (Allerberger and Dierich, 1992). The resistance rate of human LM in China remained low, similar to those reported in studies in other countries (Bertrand et al., 2016; Hansen et al., 2005; Morvan et al., 2010; Safdar and Armstrong, 2003).
High susceptibility of LM was also found among food isolates, and ST9 and ST155 were the dominant resistant genotypes (Yan et al., 2019; Zhang et al., 2019). No β-lactam resistance genes were detected, which was reported in a previous study (Shi et al., 2021). Trimethoprim in combination with sulfamethoxazole is generally used in cases of intolerance of β-lactams (Kovacevic et al., 2013), and only 3 isolates were resistant to trimethoprim/sulfamethoxazole.
Some studies showed that the most common mechanism of macrolide resistance is due to the presence of erm genes (Wilson et al., 2018); however, erm (A), erm (B), and erm (C) were not observed in this study.
Clinical LM isolates showed high genetic heterogeneity. There is an overwhelming preponderance of serogroups IIb and IIa among the clinical isolates, reflecting that food chains offer suitable niches for these LM isolates to form biofilms and survive. A previous study showed that CC1, CC6, CC2, and CC4 were more virulent and strongly associated with clinical origin (Maury et al., 2016).
However, in China, CC87 and CC8 were the dominant clones, and other studies also confirmed the prevalence of CC87 in China (Wang et al., 2019). Data from Canada and a previous study in China also showed a predominance of CC8 (Anwar et al., 2022; Knabel et al., 2012). There is clear evidence for CC6 as an emerging group of outbreaks (Bergholz et al., 2018; Gillesberg et al., 2016; Koopmans et al., 2017; Smith et al., 2019), whereas only 3 CC6 human isolates were identified in this study.
LIPI-4 was recently described as a cellobiose-family phosphotransferase system involved in neural and placental infection, which was demonstrated to distinguish hypervirulent and hypovirulent clones (Maury et al., 2016). It was detected in all CC87 and CC4 isolates, in agreement with previous findings (Painset et al., 2019; Wang et al., 2019), and in all the ST619 and ST382 isolates observed in this study. CC87 and ST619 are the most prevalent hypervirulent clones from human isolates in China and seldom associated with human listeriosis in other countries.
Broad study data suggested that CCs had a clear association with food matrices: CC1 was strongly associated with dairy products, CC121 and CC9 were associated with meat products, CC21 was associated with vegetables, and CC101 was associated with sushi (Cabal et al., 2019; Maury et al., 2019). In general, the prevalence of different CCs in different geographic regions depends largely on the types of food business operations and national consumption preferences.
CC87 was frequently isolated from Chinese salads, cooked meat products (Li et al., 2020), and seafood (Zhang et al., 2020); however, more imported and domestic food-origin isolates need to be analyzed and correlated with human origin according to WGS-based source attribution (Shen et al., 2022; Song et al., 2022; Zhang et al., 2022). Hypovirulent isolates accounted for 76.4% of isolates, which suggested that the infective dose and other virulent factors combined with certain population groups at higher risk could be important for severe infection.
As the long and variable incubation period (from a few days to several weeks) leads to recall bias and uncertain food histories, it can be very challenging to investigate and identify the source of listeriosis infection (Angelo et al., 2016). Listeriosis cases are frequently associated with consumption of contaminated RTE food products (Hazards et al., 2018).
Combined with epidemiologic and product trace-back data, limits of LM in 3 food categories—frozen drinks, RTE fruit and vegetable products, and aquatic products—were added to the National Food Safety Standard Pathogen Limits for Prepackaged Food (GB29921-2021) (NHC, 2021). The limit of LM in frozen drinks was originally set to 100 colony-forming units/g(mL), but considering the popsicle-related listeriosis case, the limit was finally set to not detectable at 25 g/mL. Strict legislation governs the limits of pathogens in food and thus minimizes the likelihood of illness emerging from high-risk food and protects the health of consumers.
WGS analysis can afford high discrimination and acquisition of a complete understanding of isolate genetic variation, so it is increasingly used in outbreak detection and investigation in the world (Jackson et al., 2016; Kwong et al., 2016; Liu et al., 2022; Moura et al., 2017; Schmid et al., 2014). The two principal approaches for strain discrimination, cgSNP and cgMLST, show concordant phylogenetic relationships, and the results are most useful when interpreted in the context of epidemiological data (Jackson et al., 2016).
In China, we integrated the cgMLST approach into routine mandatory foodborne disease surveillance and cluster detection because a standardized nomenclature system can maintain interlaboratory comparability. The SNP approach is highly discriminatory, but the choice of reference genome can significantly influence the results (Henri et al., 2017). According to the Institut Pasteur criteria (Moura et al., 2016), a cluster in cgMLST will be determined when at least 2 isolates from patients have the same CTs in TraNet.
Hypervirulent clones colonize and invade intestinal tissues better than hypovirulent clones, reflecting their adaptation to the host environment. Conversely, hypovirulent clones are adapted to food processing environments, with a higher prevalence of stress resistance genes. As indicated, WGS can not only act as an epidemiological tool to identify highly related isolates but it can also be used to detect the presence of pathogenicity islands associated with hypervirulence or particular modes of pathogenesis (such as the ability to invade the central nervous system) (Maury et al., 2016).
Conclusions
In summary, listeriosis cases remain largely undiagnosed and underreported in China. However, LM has been found to be one of the pathogens causing blood infection, central nervous system infection, and perinatal infection complications. Our results provide crucial insights into the predominance of CC87 of lineage I and CC8 of lineage II in clinical isolates.
These findings contribute to a better understanding of the genomic diversity and virulence profiles of LM isolates in China. Integration of the WGS analysis, epidemiological investigation, and source attribution will help us devise targeted interventions and make policy decisions.
Footnotes
Acknowledgments
The authors thank the Institut Pasteur teams for curation and maintenance of BIGSdb-Pasteur databases at
Authors' Contributions
W.L. was involved in conceptualization, data curation, and writing—original draft of the manuscript. Y.G. was involved in methodology and writing—review and editing. Q.C. was involved in software and visualization. X.M., X.Z., H.C., X.Y., W.C., X.L., B.Y.X., M.C., B.Y., and W.C. were involved in acquisition of data and strains and investigation. H.H. was involved in statistical analysis and validation. J.L. was involved in validation and supervision. L.Z. was involved in conception and design of the study, resources, and project administration.
Disclosure Statement
No conflict of interest exits in the submission of this manuscript, and manuscript is approved by all authors for publication.
Funding Information
This work was supported by the Ministry of Science and Technology of the People's Republic of China (Grant No. 2021YFF0703804) and Guangxi Natural Science Foundation (Grant No. 2018GXNSFAA050039).
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.