
Abstract
Listeria monocytogenes is a foodborne pathogen. In 2022, we collected 15 strains of L. monocytogenes isolated from patients in some foodborne disease sentinel monitoring hospitals in Sichuan Province. Through whole genome sequencing (WGS), we obtained the virulence genes carried by the strains, multi-locus sequence typing (MLST), core genome MLST (cgMLST), clonal complex (CC), and serum groups and constructed a phylogenetic tree and minimum spanning tree with nonhuman strains. An analysis shows that all 15 strains of L. monocytogenes carry virulence genes LIPI-1 and LIPI-2, whereas the carrying rates of LIPI-3 and LIPI-4 virulence genes are relatively low. The MLST typing results showed a total of 10 sequence types (ST), including 10 CCs, with ST7 being the dominant type. The cgMLST clearly distinguishes strains of different lineages and CC types. The serum group is divided into three types: IIa, IIb, and IVb, with IIa being the dominant serum group. An analysis of antibiotic genes showed that all 15 strains carried FosX, lin, mprF, and norB with high carrying rates. The minimum inhibitory concentration results indicated that all were susceptible to eight antibiotics (ampicillin, penicillin, tetracycline, meropenem, erythromycin, vancomycin, ciprofloxacin, and trimethoprim–sulfamethoxazole). The analysis of strains isolated from different sources of Listeria revealed varying degrees of diversity, and the contamination of meat and environment within the province is closely related to clinical cases. L. monocytogenes isolated from clinical cases in Sichuan Province carry multiple virulence and antibiotic genes, with high potential pathogenicity. It is necessary to further strengthen the monitoring and control of food and environment by L. monocytogenes within Sichuan Province.
Introduction
Listeria monocytogenes is a Gram-positive (GP) bacterium commonly found in natural environments. It possesses unique characteristics compared with other pathogens, as it can thrive in conditions of low temperature, osmotic stress, and nutrient limit (Chaturongakul et al., 2008). L. monocytogenes also exhibits relative resistance to acidity and high salt concentrations and has the ability to form biofilms, and L. monocytogenes can persist in food for extended periods (Freitag et al., 2009; Koutsoumanis et al., 2004; Lee et al., 2014). The first notable outbreak of L. monocytogenes in Canada highlighted its role as a foodborne pathogen (Schlech et al., 1983). The bacterium was initially isolated from patients in 1929. Listeriosis, caused by L. monocytogenes, is one of the most hazardous zoonotic diseases transmitted through food. Although ingesting high doses of millions of bacteria manifests as mild gastroenteritis in most individuals, it can progress to systemic infections, such as encephalitis, meningitis, gastroenteritis, septicemia, pregnancy complications, and other severe conditions, in pregnant women, the elderly, and those with compromised immune systems, even when exposed to low levels of food contamination (Al-Gburi, 2020; Charlier et al., 2017). L. monocytogenes infection during pregnancy can lead to miscarriage, stillbirth, premature delivery, and other serious consequences (Haagsma et al., 2013; Radoshevich and Cossart, 2018). Unborn babies and newborns can become infected through vertical transmission from the mother or by bacterial colonization during birth through the vaginal route (Frøen et al., 2011). In daily life, humans often contract L. monocytogenes infections by consuming raw food products or through cross-contamination with other foods. However, L. monocytogenes infections in humans are relatively rare. This study performed whole genome sequencing (WGS) on 15 L. monocytogenes strains isolated from patients and collected from sentinel monitoring hospitals for foodborne diseases in Sichuan Province in 2022. The analysis primarily focused on drug resistance genes, virulence genes, serogroup, multi-locus sequence typing (MLST) molecular typing, core genome MLST (cgMLST), and comparative analysis with nonhuman strains. The comprehensive analysis of strain sequence types was conducted, including molecular tracing, to provide data for understanding the sources of L. monocytogenes infections in patients. The research results hope to provide basic data for establishing a L. monocytogenes monitoring program and lay a foundation for further in-depth research on the molecular mechanisms of the pathogenicity and drug resistance of this bacterium.
Materials and Methods
Strains
In 2022, a total of 15 suspected strains of L. monocytogenes were collected from some areas of Sichuan Province (Chengdu, Dazhou, Guangyuan, Leshan, Panzhihua, and Yibin), which were reviewed and confirmed by our laboratory according to the national food safety standard “Food Microbiology Test—Listeria monocytogenes Test” GB4789.30-2016: inoculate the suspected strains onto Listeria chromogenic agar, incubate at 36 ± 1°C for 24 h, select typical colonies, inoculate them onto nutrient agar plates for purification, select colonies, and then identify them using VITEK 2 GP identification card (Table 1).
Patient Age and Sex, Regional Distribution of Strains, and Source Statistics
Reagents and instruments
The reagents and instruments that were used include Listeria chromogenic agar (Shanghai Xinzhong Company);VITEK 2 GP bacteria identification card (BioMérieux); nucleic acid extraction kit (Beijing Tiangen Company); VITEK 2 fully automatic microbial identification and drug sensitivity analyzer (BioMérieux); and AST Panel for Aerobic Gram Positive bacilli (Shanghai Fosun Diagnostics Technology Co., Ltd.).
WGS
Whole genome DNA was extracted from TIANamp Bacteria DNA Kit (Tiangen Biotech). Illumina platform was used to sequence the samples, and a series of data processing was carried out on the possible contamination, low-quality sequences, and joint sequences in the original sequencing data. It mainly included removing reads with 5 bp of ambiguous bases, removing reads with 20 bp of low quality (≤Q20) bases, removing adapter contamination, and removing duplicated reads. Denovo was used to sequence the bacterial genome and assemble it from scratch to obtain the optimal contig.
Virulence gene analysis
The spliced genomic data was uploaded to the BIGSdb-L. monocytogenes database (https://bigsdb.pasteur.fr/listeria/) for Listeria pathogenicity islands (LIPI), and its associated virulence genes were analyzed.
MLST and cgMLST typing
MLST analysis was performed by sequencing seven housekeeping genes (abcZ, bglA, cat, dapE, dat, ldh, and lhkA) (Ragon et al., 2008). Alleles and sequences types (STs) were determined by a comparison with allelic profiles for L. monocytogenes in the MLST database. The WGS raw data were imported into BioNumerics software and then uploaded to the National Molecular Tracing Network for Foodborne Diseases Surveillance (TraNet) calculation engine at Aliyun for de novo assembly. The cgMLST scheme included 1748 loci for L. monocytogenes in BioNumerics (Ruppitsch et al., 2015).
Polymerase chain reaction serogroup
The assembled strain contigs were uploaded one by one to BIGSdb-L. monocytogenes. Then they were compared and typed based on the five polymerase chain reaction (PCR) serogroups (IIa, IIb, IIc, IVa, and IVb) in the database (Hyden et al., 2016).
Drug resistance genes’ analysis and minimum inhibitory concentration assays
The spliced genome data were uploaded to the comprehensive antibiotic resistance database (CARD; https://card.mcmaster.ca/analyze/rgi), Resfinder (http://genepi.food.dtu.dk/resfinder), and BIGSdb-L. monocytogenes to obtain antibiotic resistance genes in the strain genome sequence.
We used AST Panel for Aerobic Gram Positive bacilli for minimum inhibitory concentration (MIC) testing according to the Clinical and Laboratory Standards Institute (CLSI) in the United States. The test plate was equipped with a series of antibiotic reagents with double dilution concentrations for eight antibiotics (ampicillin, penicillin, tetracycline, meropenem, trimethoprim–sulfamethoxazole, erythromycin, vancomycin, and ciprofloxacin), and ATCC 29213 Staphylococcus aureus was used as the quality control strain.
Comparison and analysis with nonhuman strains
In order to analyze the potential contamination sources of the clinical case, a total of 41 L. monocytogenes (including food and environmental isolates from multiple provinces including Sichuan, Guangdong, Beijing city, Heilongjiang, Neimenggu, Henan, Hunan, Jilin, Shanxi 1, Shanxi 2, Zhejiang, Jiangxi, Shanghai, and Hubei in China) were downloaded from the NCBI Pathogen database for further analysis. The minimum spanning tree analysis was conducted using BioNumerics software to compare these 27 nonhuman strains with 15 clinical strains in this study.
Data processing and plotting
The exported data were organized using Excel and plotted using BioNumerics 7.6cn and GraphPad Prism 9.0 software.
Results
Virulence gene analysis
Fifteen strains were found to carry prfA, plcB, and mpl (belonging to LIPI-1) and inlA, inl, and inlC (belonging to LIPI-2), one strain did not carry plcA and actA, three strains carried LIPI-3, and two strains carried LIPI-4. One strain carried both LIPI-3 and LIPI-4 (Fig. 1).
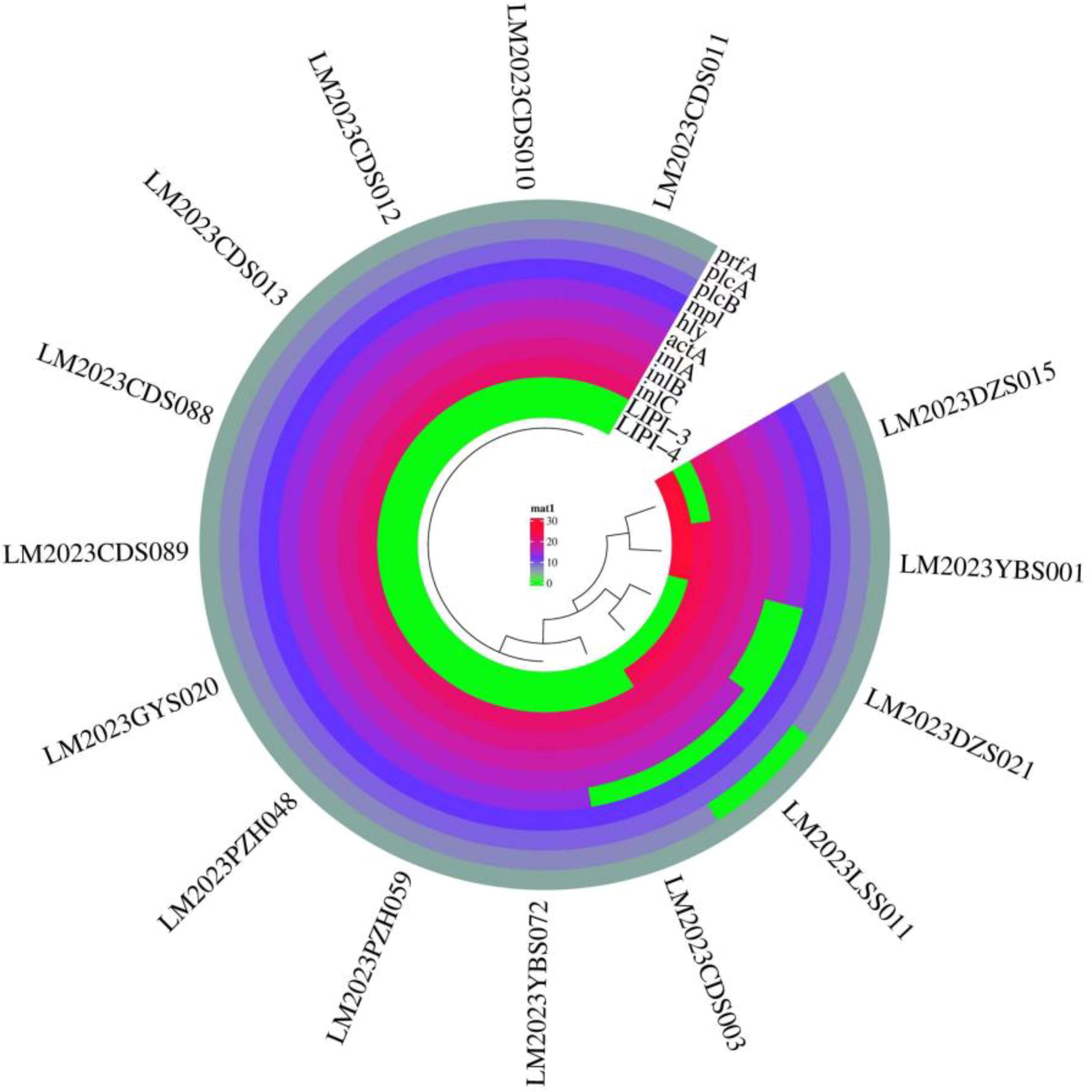
Ring clustering diagram of virulence genes. The green highlight indicates a deletion of virulence genes.
cgMLST, MLST, and clonal complex typing
According to the cgMLST homology criteria, there is only one allele difference among the three strains from Chengdu. The two strains from Dazhou City and Leshan City were also determined to be homologous with a difference of four alleles, and the others were not homologous. The 15 strains were classified into 10 sequence types (ST), including 10 clonal complexes (CC), as shown in Figure 2. ST7 (CC7) was the dominant type with five strains, followed by ST3 type with two strains, and the other eight strains were different in types. They are ST5 (CC3), ST8 (CC8), ST87 (CC87), ST451 (CC11), ST378(CC19), ST2 (CC2), ST619 (CC619), and ST736 (CC736). ST3, ST2, ST619, ST87, ST736, and ST5 belong to lineage I. ST7, ST451, ST378, and ST8 belong to lineage II. In addition to ST451 belonging to CC11 and ST378 belonging to CC19, the ST type and CC type numbers of the other 13 strains were consistent (Fig. 2).

Phylogenetic tree of 15 L. monocytogenes isolates based on cgMLST. The corresponding data, including the name of the isolates (strain ID), Cities, sample source MLST type (ST), MLST clonal complexes (CC), and serogroups, were shown alongside the dendrogram to the right. cgMLST, core genome MLST; MLST, multi-locus sequence typing; ST, sequence type; wgMLST, whole genome MLST.
Lineage and PCR genoserogroup
In this study, IIa is the dominant serum group, belonging to lineage II. There are eight strains of this serotype, followed by IIb serum group, belonging to lineage I with six strains. Only one strain belongs to the IVb serum group, belonging to lineage III (Fig. 2). IIa, IIb, IIc, and IVb are all dominant types in China. Except for IIc, the other three account for the majority of human clinical cases of L. monocytogenes (Wang et al., 2015), which are rare in literature reports (Fig. 2).
Antibiotic genes and antimicrobial resistance
In this study, four antibiotic genes FosX, lin, mprF, and norB were mainly detected, and all strains carried FosX and lin, which were related to the resistance to phosphonic acid antibiotics and lincosamide antibiotics, respectively. The resistance mechanism was to inactivate antibiotics, and 12 strains carried mprF, which mainly changed the target of peptide antibiotics, resulting in resistance. Six strains carried norB, which is associated with fluoroquinolone antibiotic resistance through antibiotic efflux pumps. Three strains carried all four resistance genes.
According to CLSI, 15 strains were found to be susceptible to eight antibiotics (ampicillin, penicillin, tetracycline, meropenem, erythromycin, vancomycin, ciprofloxacin, and trimethoprim–sulfamethoxazole). Vancomycin belongs to peptide antibiotics, whereas ciprofloxacin belongs to fluoroquinolone antibiotics. Although carrying corresponding resistance genes, the resistance phenotype is not consistent with it (Table 2).
Minimum Inventory Concentration Test Results
Refers to EUCAST; others refer to CLSI M45-A.
CLSI, clinical and laboratory standards institute; EUCAST, european committee on antimicrobial susceptibility testing.
Comparison and analysis with nonhuman strains
The cgMLST of 4 clinical and 14 food isolates from Sichuan correctly grouped these isolates together. Among these food strains, 6 isolates were ST87 (from Chengdu, Yan, Yibin, and Mianyang), with a maximum allele difference of 1–9 between them, and a clinical strain from Dazhou (SC2022DZS015), and 5 were ST378 (from Chengdu, Yibin, and Ziyang), and their allele difference with a clinical strain from Chengdu (SC2022CDS088) was 1–5. There were 2 food isolation strains from Chengdu, ST619, with only 2 allele differences from the clinical strain SC2022YBS001 in Yibin and 1 food isolation strain from Mianyang, ST5, with 12 allele differences from SC2022CDS003. These isolates were cultured from raw food products (meat) in 2022. It can be clearly seen that the 4 clinical isolates match very well with the strains isolated from meat (Fig. 3A). In this study, a clinical strain had 10 allele differences from an environmental isolation strain in Sichuan Province, 17 allele differences from a food strain in Guangdong Province, and more than 1000 allele differences from food strains in other provinces (Fig. 3B).
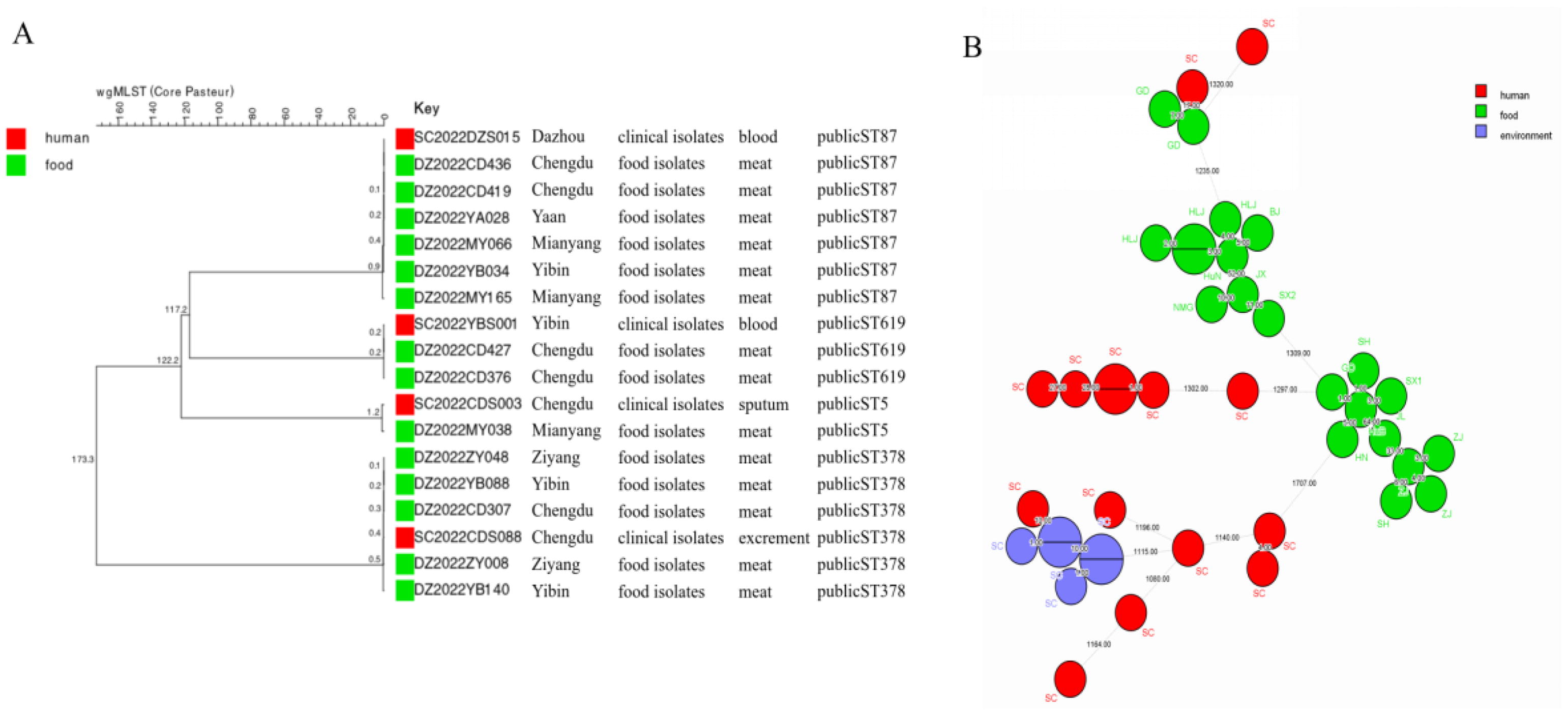
Comparison of clinical strains and nonhuman strains.
Discussion
L. monocytogenes is widely present in nature, and human listeriosis is often caused by ingestion of food contaminated with L. monocytogenes. To understand the characteristics of L. monocytogenes strains with high pathogenicity potential in food colonization, pathogen survival, and virulence gene resistance, this study used WGS technology to mine and analyze the genomic features of L. monocytogenes, aiming to provide data support for establishing a more comprehensive foodborne disease monitoring system.
Listeria pathogenicity island (LIPI) is considered to be an unstable chromosomal region that carries genes related to specific metabolic activities, antibiotic resistance, or pathogenesis and can be transferred horizontally between bacteria (Gal‐Mor and Finlay, 2006). LIPI-1 consists of six virulence genes, prfA, plcA, hly, mpl, actA, and plcB, which are closely related to L. monocytogenes invasion of host, intracellular proliferation, intercellular transfer, diffusion, and intracellular escape. In this study, only one strain was deficient in plcA, and three strains were deficient in hly. prfA, mpl, and plcB carry rate is 100%; inlA, inlB, and inlC of LIPI-2 are the internal genes required for the internalization of L. monocytogenes in host cells, which play a key role in the bacterial adhesion and invasion process. All 15 strains in this study carried these three internal genes. In addition, there were three strains carrying LIPI-3 and two strains carrying LIPI-4. LIPI-3 assisted L. monocytogenes to cross the black bowel barrier and contained eight genes: llsA, llsG, llsH, llsX, llsB, llsY, llsD, and llsP. LIPI-4 is a phosphotransferase system belonging to the cellobiose family, which, in addition to its role in crossing the blood–brain barrier, can also enhance the toxicity of L. monocytogenes to the nerves and placenta (Chen et al., 2020a). In addition, the overall carrying rate of bsh, ami, and aut is relatively high, and they mainly participate in the bacterial adhesion and invasion process (Cabanes et al., 2002; Shi et al., 2021). Overall, the virulence gene carrying rate of clinical isolates was high, suggesting that the pathogenicity might be stronger than other isolates.
There are three strains of bacteria with hly deficiency, two of which are ST3 and one is ST5. The strains with plcA deficiency and actA deficiency are ST3, one carrying all virulence genes is ST619, one carrying LIPI-4 but not LIPI-3 is ST87, and two strains carrying LIPI-3 but not LIPI-4 are both ST3, suggesting that ST3, ST87, and ST619 were highly virulent strains. The results were consistent with the analysis and study of Listeria cases in Henan by Chui et al. (2021). In the study by Huo et al. (2017), the three types ST5, ST8, and ST87 were mixed with human and foodborne strains. Among them, ST378 existed only in clinical isolates, and this study also found one case of this type. The proportion of PCR serogroup related to human infection, including IIa, IIb, and IVb, is relatively high (Chen et al., 2020a). The results of this study are consistent with those of Wang Yan et al. from 28 clinical isolates (Wang et al., 2015). According to the report by Li et al. (2018), the average detection rate of L. monocytogenes in food from 28 provinces in China was 4.42%, and the detection rate of meat products was the highest. ST9 and ST8 are dominant sequence types in food, whereas ST3, ST8, and ST87 are the most important sequence types clinically, and these three types were detected in this study. In 2017, the L. monocytogenes strains detected in ready-to-eat foods in China were mainly serogroup IIa, with CC8, CC101, and CC87 as the dominant CC types. The strains carrying LIPI-4 were mainly the endemic strain CC87 in China, most of which were isolated from Chinese cold dishes and ready-to-eat fruits and vegetables (Li et al., 2020), and the L. monocytogenes isolates in this clinical case had similar molecular characteristics. This suggests that ingestion of ready-to-eat foods contaminated with these types of L. monocytogenes may increase the risk of the disease. It is worth noting that CC87 has been identified as the most common subgroup associated with both food and human clinical infections in China. ST87 belongs to CC87, which has been identified as a prevalent sequence type of L. monocytogenes of different origins in China (Chen et al., 2020b; Wang et al., 2015). The three strains isolated from blood, sputum, and amniotic fluid in Chengdu have only one allele difference and have been identified as homologous. WGS results confirmed that the infection of the newborn was mainly caused by vertical transmission from the mother. In addition, two strains from Dazhou City and Leshan City were determined to be homologous with a difference of four alleles. Therefore, the cgMLST typing method has great advantages for surveillance and outbreak identification of foodborne diseases in Sichuan.
Scortti et al. (2018) reported that a FosX enzyme encoded in the Listeria core genome confers intrinsic fosfomycin resistance to Listeria spp. and carried the FosX gene, which was consistent with this study. Fifteen strains of L. monocytogenes mainly carried four resistance genes, FosX, lin, mprF, and norB, and the resistance mechanism included antibiotic inactivation, antibiotic target change, and antibiotic effluvium pump. But all strains were susceptible to vancomycin and ciprofloxacin, indicating that carrying resistance genes may not necessarily have a resistance phenotype. We can determine the presence of antimicrobial resistance genes through WGS as a clue, but whether resistance is present or not still requires confirmation of the resistance phenotype. At present, the L. monocytogenes strains isolated from patients are limited and have not been traced in a timely manner. Comparison of L. monocytogenes strains isolated from different provinces and sources revealed that clinical isolates in Sichuan are closely related to the contamination of meat products and the environment within the province. Therefore, prevention and control of the spread of L. monocytogenes from food and the environment to humans should be implemented. Therefore, WGS, a high-resolution source of infection-tracking technology, can be used in a timely manner to investigate the potential food sources of maternal L. monocytogenes infection and the causes of the infection in patients from different regions and cities. This is of great significance for further prevention, diagnosis, and treatment of L. monocytogenes.
Footnotes
Data Availability Statement
Authors’ Contributions
X.Y. conceived and designed research. Q.L., W.H., T.X., L.Z., G.L., and H.L. performed experiments and acquired, interpreted, and analyzed the data. X.Y. and Q.L. wrote the article. All authors read and approved the submitted version.
Disclosure Statement
The authors declare no conflict of interest in relation to this study.
Funding Information
This work was supported by the Sichuan Science and Technology Program (grant no. 2022ZDZX0017).
Supplementary Material
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.