
Abstract
Twenty-one environmental Vibrio vulnificus strains from the Centro de Investigación Científica y de Educación Superior de Ensenada (CICESE) bacteria collection, 20 of them isolated from oyster samples and 1 from a reported clinical case, were sequenced to analyze the genomic divergence between 2 genotypes, E-genotype and C-genotype, proposed by various groups to distinguish clinical (C) from environmental (E) V. vulnificus strains. As indicated in previous analyses of PFGE, MLST, and rtxA, 9 of the CICESE isolates were identified as vcgE, compared with 12 as vcgC. Separation of the genotypes into these 2 groups was confirmed in this study, based on the presence of certain genes in the 21 genomes, the presence of virulence factors, and rtxA sequencing. Most genomes from oyster isolates expressed rtxA-C type, with the exception being rtxA-M type detected in CICESE-594 a vcgE strain isolated from a clinical case. Although several genetic approaches clearly indicate differences between the C- and E-genotypes, none of them, including those in this study, can highlight a single factor that could be used to indicate the potential pathogenicity of V. vulnificus isolated from oysters.
Introduction
Vibrio vulnificus is an opportunistic pathogen that can cause gastroenteritis, necrotizing fasciitis, and primary septicemia, most cases of which have been associated with seafood consumption (Baker-Austin and Oliver, 2018); the mortality rate in the United States due to septicemia is >50% (Jones and Oliver, 2009). A risk assessment report by the Food and Agriculture Organization of the United Nations (FAO/WHO, 2005) has noted that the virulence of V. vulnificus is multifaceted, prompting them to propose that all V. vulnificus strains must be considered virulent. However, Bier et al. (2013) suggest that despite the frequent detection of V. vulnificus, the number of cases that have been reported in the Baltic Sea region was low.
Based on genetic polymorphisms of the virulence-correlated gene (vcg), V. vulnificus strains are subtyped into two groups that are linked to their origin: vcgC for those that have a clinical source and vcgE for those that are isolated from the environment (Rosche et al., 2005 and Rosche et al., 2010). In one study, Guerrero et al. (2019) have reported that strains that were isolated from oyster samples presented both genotypes and that C-genotype strains from oysters are genetically associated with reference C-genotype strains that originate from clinical cases. These authors concluded that not all strains isolated from oyster samples belong to the E-genotype. In another study, Rosche et al. (2010) have reported that the frequency of infection in at-risk individuals is lower when they are exposed to strains with the vcgE versus vcgC genotype. Thiaville et al. (2011) noted that, in a mouse model, mortalities were registered for both genotypes, concluding that genotype correlated with, but did not strictly predict, virulence in Biotype 1 and pointed out the need for further studies to identify virulence genes as markers of virulence. Differences between the C-genotype and E-genotype have been described using several approaches, including 16S rRNA gene analysis (Nilsson et al., 2003; Vickery et al., 2007; Wood and Arias, 2012), multilocus sequence typing (Bisharat et al., 2020), rtxA gene cluster (Kwak et al., 2011), genomic analysis (Morrison et al., 2012), and/or oligonucleotide probes (Aznar et al., 1994).
The pathogenicity of V. vulnificus is linked to several virulence factors, such as metalloprotease (VvpE), capsular polysaccharide (CPS), and the regulation of iron acquisition (Jones and Oliver, 2009). However, cytolysin–hemolysin (VvhA) and multifunctional autoprocessing RTX toxin (MARTXVv), encoded by vvhA and rtxA, respectively, are considered the main virulence factors (Jeong and Satchell, 2012; Lee et al., 2018; Lee et al., 2007; Lo et al., 2011).
The aim of this study was to identify key genetic characteristics among the genomes of 21 vcgE and vcgC strains, most of which (20) were isolated from oyster samples and one from a human clinical case (Bier et al., 2013). The study focused on phylogenetic relatedness and the presence of virulence factors as an indication of the potential virulence of V. vulnificus strains isolated from oyster samples.
Materials and Methods
Genomic sequencing
Twenty-one V. vulnificus strains (Table 1), most of which (20) were isolated from oyster samples collected during 2 years (2012–2013) at the principal seafood retail market in Mexico City, were classified as vcgC or vcgE (Guerrero et al., 2019; Guerrero et al., 2015). One vcgE strain Centro de Investigación Científica y de Educación Superior de Ensenada (CICESE)-594 (ATCC 27572T), isolated from a human blood sample (Bier et al., 2013), was included in the study as a reference vcgE strain isolated from a clinical case. All the strains were sequenced to determine the genomic differences between genotypes. A single colony from each strain was cultured in Zobell marine agar (Oppdenheimer and ZoBell, 1952) at 37°C overnight. Genomic DNA was extracted using the Wizard Genomics DNA™ purification kit (Promega Corporation, Madison, WI) according to the manufacturer’s instructions. The resulting DNA was maintained at −20°C until use. Libraries for each strain were created using TruSeq Sample Preparation and then applied to whole-genome sequencing, performed on a MiSeq system (Illumina, San Diego, CA) at CICESE, Ensenada, Baja California, Mexico.
RAL, reads average length (bp); Contigs, number of contigs ≥200 bp; GS, genome size (Mb); GC, GC content (%); CS, number of detected coding sequence.
Genome assembly
The obtained reads were used to determine the coverage depth using the coverage.sh script (https://github.com/cabraham03/coverage/blob/main/coverage.sh) with Samtools V1.2 (Li et al., 2009) and BWA-MEM V0.7.12 (Li and Durbin, 2009); the resulting metrics were visualized in Qualimap (García-Alcalde et al., 2012). Each genome was assembled in SPAdes V3.8 (Bankevich et al., 2012), and the de novo genome assembly was performed in careful mode with the -k 21,33,55,77,99,127 parameters.
Pan- and core-genome analyses
Pan- and core-genome plots were constructed, based on shared genes of the 21 CICESE genomes. The analyses were performed as follows: contigs of each genome were used to obtain the predicted amino acid in Prokka V1.14.6 (Seemann, 2014), the generated files (gff) were used with Roary V3.12.0 (Page et al., 2015), with 90 as the minimum percentage of identity, and the resulting matrix of the presence/absence of genes was used to generate the pan and core genomes using ggplot2 V3.4.0 (https://github.com/tidyverse/ggplot2). The phylogenetic tree and heatmap in Figure 1 were constructed with genes of the core genome (3462 genes), obtained by Roary analysis, by calculating the distance by unweighted pair group method with arithmetic mean (UPGMA) in phangorn V2.10 (https://github.com/KlausVigo/phangorn) and plotted in ggtree V3.6.2 (https://github.com/YuLab-SMU/ggtree; Yu et al., 2017) using R V4.1.1, implemented in the RStudio V2021.09.0 environment.
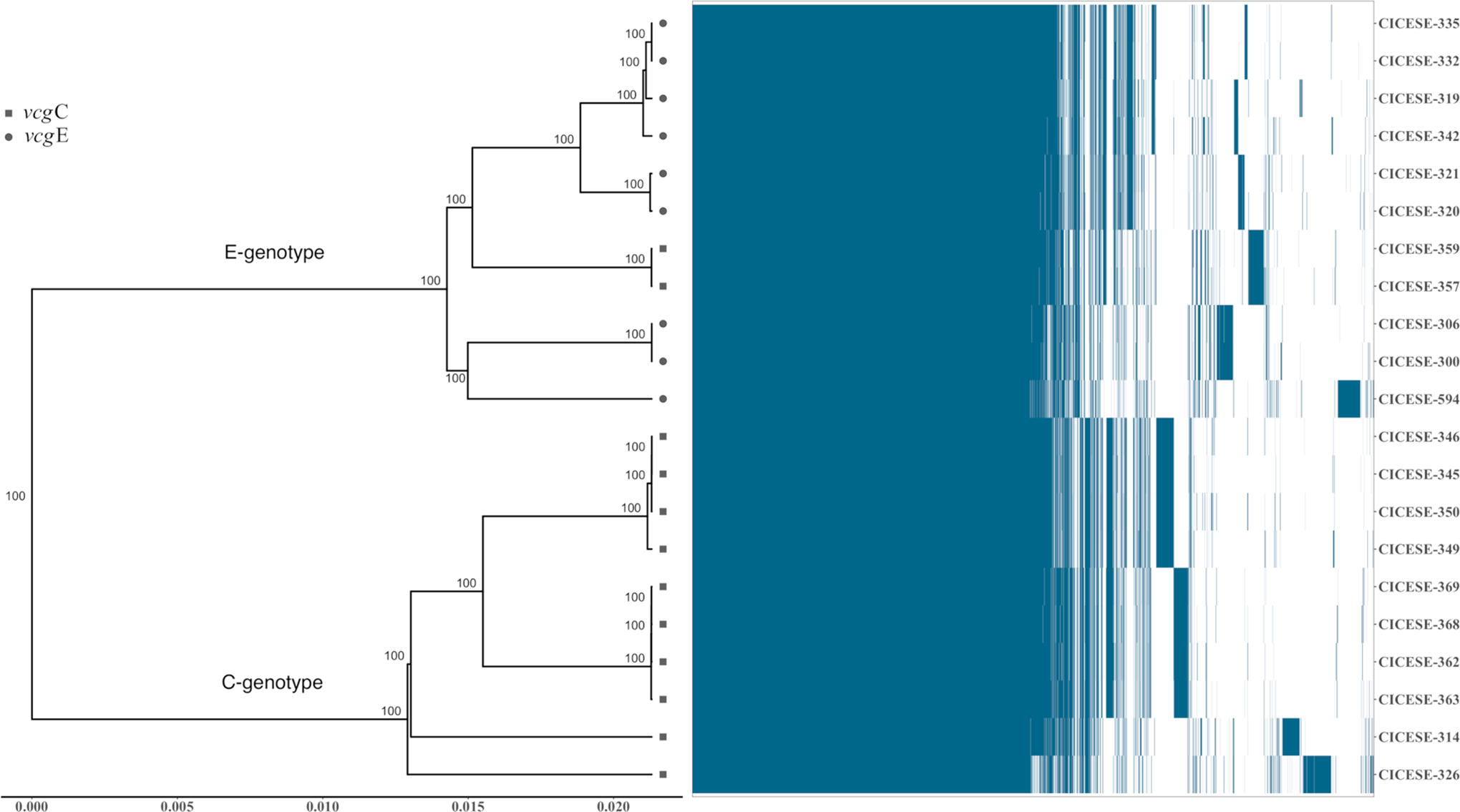
Phylogenetic tree and heatmap generated based on the presence/absence of genes among the 21 genomes. The presence and absence of genes are indicated in blue and white, respectively. The vcgE strains are indicated by a circle (●) and the vcgC strains are denoted by a square (■).
Virulence factors
The presence of virulence factors that were encoded in the genomes was inferred by comparing the predicted genes (nucleotides) of each genome against the Virulence Factor Database (VFDB, http://www.mgc.ac.cn/VFs/; Liu et al., 2019). Because some alleles that are associated with the rtxA-M type were not reported in the database, we added the sequences for rtxA (VVMO6_RS19590), rtxB (VVMO6_RS19605), rtxC (VVMO6_RS19595), rtxD (VVMO6_RS19610), and rtxE (VVMO6_RS19615), which have been reported for the rtxA-M type MO624/O V. vulnificus reference genome (NC_014966) in the VFDB. The analyses were performed using a local search with BLAST V2.2.29+ [≥85% identity (pident), ≥85% query coverage (qcovs), and ≥85% of the relation of alignment length/slen], implemented against the VFDB.
The total detected virulence factors (157) in the 21 genomes were aligned with DECIPHER V2022.0 (https://github.com/news-r/decipher) and then concatenated for each genome. The detected virulence factors were used to generate a presence/absence plot in ggplot2 and gggenes V0.4.1 (https://github.com/wilkox/gggenes). The virulence factors detected in common in both genotypes, among the 21 genomes, were used to construct a phylogenetic tree (Fig. 2) by calculating the pairwise distance by UPGMA, implemented with phangorn, and plotted with ggtree.

Phylogenetic tree generated by common virulence factors (126) and heatmap of detected genes associated with virulence factors (157) in the 21 studied genomes. The x-axis denotes the detected genes (145,906 bp). The E- and C-genotypes are indicated by a circle (●) and the C-genotype by a square (■), respectively.
Analysis of the rtxA gene
Sequences of rtx
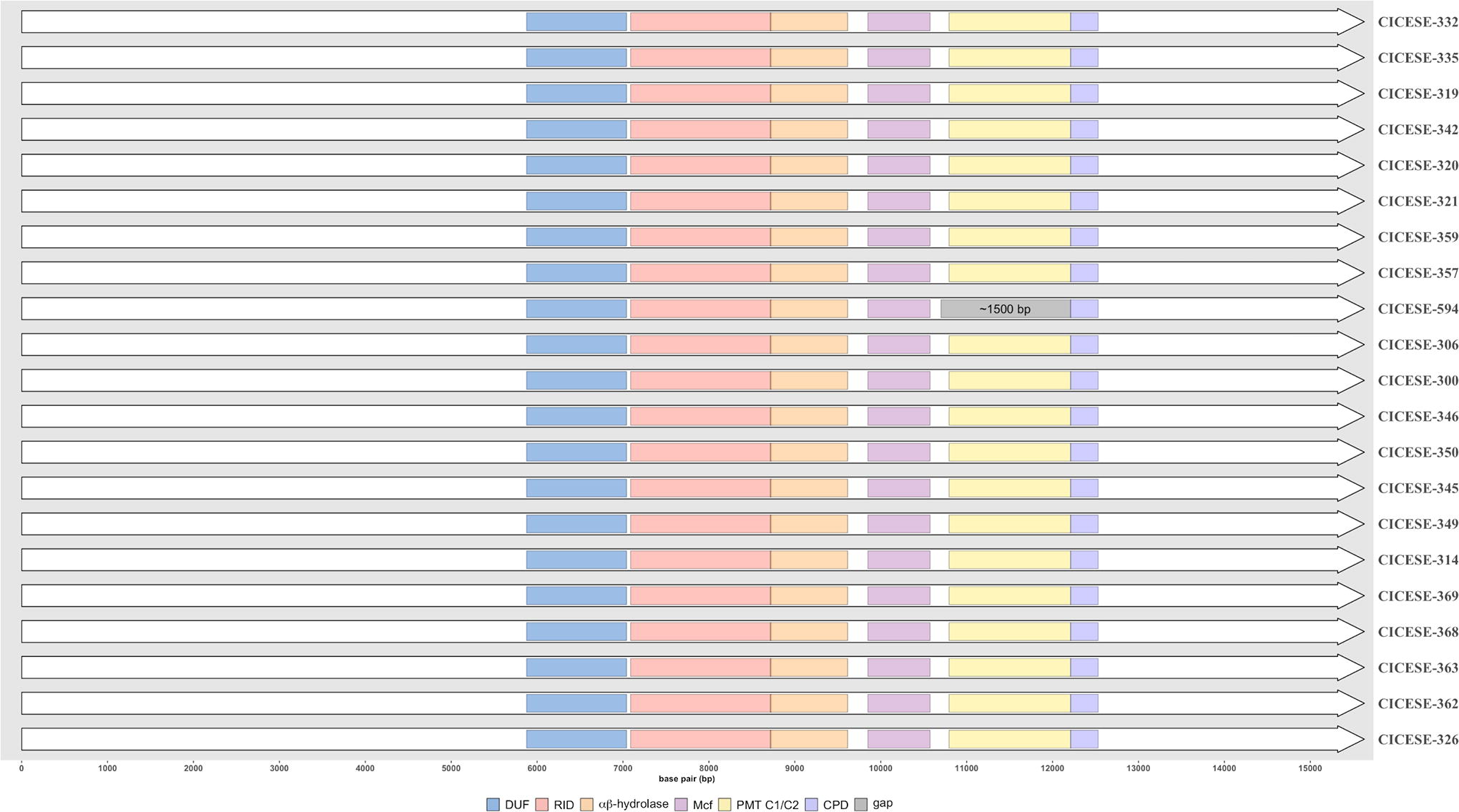
Alignment of rtxA sequences. The domains are represented with different colors and the gray zone indicates a gap of ∼1500 bp in CICESE-594.
Genomic comparison
Chromosomes I and II, presented in Figure 4, were assembled in two supercontigs on the MeDuSa web server (http://combo.dbe.unifi.it/medusa; Bosi et al., 2015) using YJ016 (GenBank accession: NC_005139 and NC_005140) as the genomic reference and then aligned with BLAST Ring Image Generator (BRIG, V0.95; Alikhan et al., 2011).

BRIG match comparison of the 21 Vibrio vulnificus genomes and the YJ016 reference genome, for chromosomes I and II; the rings are colored on a sliding scale according to percentage identity (100%, 90%, or 70%). Colors correspond to vcgE (yellow), vcgC (green), and YJ016 (blue). The figure is not drawn to scale.
The raw data for the 21 studied V. vulnificus strains, was deposited at the NCBI SRA, BioProject PRJNA970315, under accession numbers SRX23398382 to SRX23398402.
Results
The reads had an average length of between 74 pb for CICESE-300 and 224 pb for CICESE-321. The coverage depth differed between strains, the lowest of which was 15.9X for CICESE-594 and peaking at 93.6X for CICESE-349. Genomes were assembled in contigs (≥200 bp), ranging from 130 contigs (N50 = 144,317 and L50 = 11) for CICESE-306 to 242 contigs (N50 = 210,352 and L50 = 7) for CICESE-349. The remaining metrics for the 21 V. vulnificus strains (9 vcgE and 12 vcgC) are presented in Table 1.
Pan and core genome
The pan genome of the 21 genomes of V. vulnificus strains comprised 7007 genes, of which 3462 (49.4%) were identified as core genes that were shared by at least 99% of the strains, 135 were soft-core genes that were shared by 95–99% of the strains, 1523 were identified as shell genes that were shared by 15–95% of the strains, and 1887 were cloud genes that were detected in <15% of the strains. Also, 54.7% (3833) among the detected genes were annotated as hypothetical proteins.
Figure 1 shows a phylogenetic tree (UPGMA) that was generated with the core genome (3462 genes) obtained from Roary analyses and a heatmap that was constructed based on the presence/absence of genes among the 21 CICESE strains. Genomes were separated into two clusters: C-genotype, containing 10 of the 12 vcgC genomes, and E-genotype, which included all vcgE genomes plus 2 vcgC genomes (CICESE-357 and CICESE-359), which were associated with a separate group within the E-genotype cluster, as it was previously reported (Guerrero et al., 2015).
Virulence factors
Further screening of the genomes for virulence factors was conducted by local BLAST search of the 21 genomes against the VFDB. In the detected genes, 157 virulence factors were observed in the analyses, of which 126 (∼80%) were shared between the 21 genomes. Among them, the rtxA gene cluster (rtxA, rtxB, rtxC, rtxD, and rtxE) was detected in all genomes. Other common genes included cytotoxin–hemolysin (vvhA), metalloprotease (hap/vvp), type IV pilus (pilB, pilC, pilM, and pilO), MSHA type IV pilus proteins (mshB, mshD, mshF, mshG, and mshH), the Type II secretion system (epsC, epsE, epsF, and epsG), and flagellar genes (flaA, flaB, flaE, flaG, and flaI).
Type vcgC genomes harbored most of the detected genes (156), but several of them (flaC, VV1_RS15620, VV1_RS01700, VV1_RS01705, pilA, VV1_RS15585) were associated only with this type. Type vcgE genomes presented with 151 virulence factors; mshA was detected in 6 of the 10 vcgE genomes (CICESE-319, CICESE-320, CICESE-321, CICESE-332, CICESE-335, and CICESE-342). Figure 2 shows a phylogenetic tree that is based on the common virulence factors (126 concatenated genes, ∼145,906 bp). The phylogenetic differences between the 21 genomes formed 2 clusters: the C-genotype cluster comprised 10 vcgC genomes, and the E-genotype cluster contained all vcgE genomes plus 2 vcgC genomes (CICESE-357 and CICESE-359).
Analysis of the rtxA gene
In analyses that were based on the alignment of rtxA, of the 21 genomes presented with the order and structure of rtxA-C type reported for the YJ016 V. vulnificus reference strain (BJE04_RS20545, homology >93.9%), except for CICESE-594, which had a homology of >93.1% with rtxA-M type reported for the V. vulnificus MO6-24/O (VVMO6_RS19590) reference genome, the differences were associated primarily with the lack (gap of ∼1500 bp) of the PMT C1\C2 domain in CICESE-594 (Fig. 3).
Genomic comparison
Compared with the BLAST Ring Image Generator (BRIG) tool, the alignment grouped the 21 genomes into 2 chromosomes, with an expected size of ∼3.35 and ∼1.85 Mb for chromosomes I and II, respectively (Fig. 4), ordered as follows from inner to outer rings: vcgE genomes (orange) CICESE-300, −306, −319, −320, −321, −332, −335, −342, and −594 and vcgC genomes (green) CICESE-314, −326, −345, −346, −349, −350, −357, −359, −362, −363, −368, and −369, with
Discussion
Our results on the phylogenetic tree and the heatmap in Figure 1, based on the presence/absence of genes among genomes, and the phylogenetic tree in Figure 2, based on detected genes associated with virulence factors, confirm the findings from several groups, in which genetic polymorphisms in V. vulnificus strains are separated into two genotypes, E- and C-genotypes (Bier et al., 2013; Bisharat et al., 2020; Kwak et al., 2011; Morrison et al., 2012). Guerrero et al. (2015) have also indicated the separation of these two genotypes (vcgE and vcgC) by PFGE, MLST, and rtxA analysis of oyster isolates. Thus, many groups have concluded that V. vulnificus strains clearly separate into these two genotypes. Previous reports have indicated that differences in gene expression between genotypes, such as the ability to survive and grow in human serum conditions and the ability to attach, reflect disparities in life style between genotypes (Bogard and Oliver 2007; Phippen and Oliver, 2015; Smith and Oliver, 2006; Williams et al., 2014). Recently, Kling et al. (2022) and Lydon et al. (2021) studying V. vulnificus human clinical isolates with both genotypes, have concluded that vcg genotypes or the MARTX toxinotypes do not significantly correlate with a severe disease outcome.
Our results, based on the detected virulence factors, indicate that genomes from the 21 studied strains harbor 157 virulence factors, 126 of which were shared by both genotypes. These findings are consistent with previous data that indicate the existence of several virulence factors in both genotypes—primarily CPS, metalloproteases, and lipopolysaccharides (Horseman and Surani, 2011). Roig et al. (2017) identified over 250 genes that were associated with virulence factors but with strains that were isolated from different habitats and localities. Nevertheless, genes that are essential for colonization and dissemination during infection, such as the rtxA gene cluster (rtxA, rtxB, rtxC, rtxD, and rtxE) and vvhA (Gavin and Satchell, 2019; Jeong and Satchell, 2012; Kwak et al., 2011; Lee et al., 2008; Lo et al., 2011), were observed in all CICESE strains.
Strain CICESE-594 (ATCC 27562T), isolated from human blood, which was registered as vcgE and 16S rRNA type AB by Bier et al. (2013), was also associated with the vcgE cluster. In our results, most of the studied genomes presented the rtxA-C type, except the CICESE-594, that presented the M-type, having a significant gap of ∼1500 bp, where the PMT C1/C2 domain was absent. This result is notable among vcgE V. vulnificus isolates from oysters, meriting further examination. The MARTX toxin encoded by rtxA (Kwak et al., 2011) is responsible for intestinal tissue damage and inflammation (Jeong and Satchell, 2012), pore formation in eukaryotic cell plasma membranes (Satchell, 2011), and the induction of cytoskeletal arrangement (Guo et al., 2019). Although Kling et al. (2022) concluded that the multifunctional autoprocessing repeat-in-toxin (MARTX) toxinotype does not predict severity of infection, isolates with the vcgC genotype trended—albeit insignificantly so—toward having a higher proportion of the rtxA-M type of MARTX toxin.
Conclusions
This study focused on V. vulnificus strains isolated from oyster samples collected during 2 years at the main seafood retail market in Mexico City, therefore, ready for human consumption. Previous studies with the same oyster-isolated strains registered the presence of both genotypes, vcgE (environmental) and vcgC (clinical). The hypothesis in this study was to analyze the registered virulence factors, in strains isolated from oyster samples, to find differences and similarities among both genotypes as markers of virulence.
Although this study does not identify virulence genes as markets of virulence, our results confirm phylogenetic divergences between C- and E-genotypes. This study also registers that the vcgE CICESE-594 (ATCC 27572T) strain isolated from a clinical case presents a rtxA-M type that differs from the other V. vulnificus-studied strains, which presented the rtxA-C type. Nevertheless, recent studies on V. vulnificus human clinical isolates corroborate that vcg genotypes or the MARTX toxinotype do not significantly correlate with virulence and may not be an appropriate measure in seafood surveillance programs. Therefore, as FAO/WHO recommended, all V. vulnificus isolated from oysters must be considered virulent.
Data Sharing Statement
The raw data for the 21 studied Vibrio vulnificus strains were deposited at the NCBI SRA, BioProject PRJNA970315, under accession numbers SRX23398382 to SRX23398402.
Footnotes
Acknowledgments
Authors’ Contributions
M.L.L.P. and A.G.: conceptualization (lead), writing—original draft (lead), formal analysis (lead), and writing—review (lead); A.G.: software (equal). A.G., C.E.G.S., and A.V.M.V.: methodology and writing—review (lead); M.L.L.P.: project administration (lead) and resources (lead).
Disclosure Statement
The funders had no role in the study design or data collection or analysis; therefore, no competing financial interests exist.
Funding Information
Funding was provided (2009–2014) by “Fondo Sectorial Salud-CONAHCyT,” grant number S008-2009-114024 to MLLP, and CICESE internal project 682 135. Economic support for AG was provided by “Cátedras CONAHCyT-CIAD.”