
Abstract
Campylobacter jejuni (C. jejuni), a common foodborne zoonotic pathogen, usually causes gastroenteritis and rarely causes extraintestinal infections such as bacteremia. This study investigates a rare case of invasive C. jejuni bacteremia in a 23-day-old infant in Nanchang, China. Epidemiological data were collected from the infant’s cohabitants (3). The cohabitants’ anal swabs (3), milk powder (1), a feeding bottle (1), and kitchen items (3) were collected and screened for C. jejuni. Antimicrobial susceptibility testing was conducted, and whole genome sequencing was performed for genetic analysis, including whole genome multilocus sequence typing (wgMLST), single-nucleotide polymorphism, antimicrobial resistance (AMR) and virulence gene profiling. As a result, three C. jejuni strains were isolated from the infant, the infant’s grandmother, and the feeding bottle. The results revealed that the three C. jejuni isolates were clonally related, sharing minimal genetic differences. The infant’s grandmother, who had slaughtered a live chicken 4 days before the infant’s illness, was identified as the likely source of transmission. AMR profiles showed resistance to fluoroquinolones and cephalosporins. The three isolates were found to carry the blaOXA-184 gene and a chromosomal mutation in gyrA (T86I). Additionally, 69 virulence genes were identified in all isolates, including those associated with adherence (4), glycosylation system (23), motility and export apparatus (38), cytolethal toxin (3), and invasion (1). This case represents the third reported instance of C. jejuni bacteremia in an infant in China. The analysis confirmed a transmission pathway from the grandmother.
Keywords
Introduction
Campylobacter jejuni (C. jejuni) is recognized as one of the most common causes of bacterial enteric illness in humans worldwide (Caron et al., 2023). In the United States, over 1.35 million people suffer from diarrhea caused by C. jejuni annually (Centers for Disease Control and Prevention, 2024). In Europe, C. jejuni is a major pathogens of bacterial acute gastroenteritis (Botelho et al., 2022). Moreover, an overall increase in campylobacteriosis has been observed in developing countries since 2005 (do Nascimento Veras et al., 2018). Given its widespread impact, C. jejuni infection has become a global health issue. Campylobacteriosis is a zoonotic disease that humans can contract through the consumption or handling of meat contaminated with C. jejuni, with poultry being identified as the most common source of infection (Redondo et al., 2019).
C. jejuni infection usually leads to acute and self-limited gastroenteritis with fever, diarrhea, and abdominal pain. Extraintestinal infections of C. jejuni in children are rare, including bacteremia, meningitis, and osteomyelitis (Valenčak-Ignjatić et al., 2022). Bacteremia occurs in <1% of campylobacteriosis cases, mainly in immunocompromised and elderly patients (Botelho et al., 2022; Mizuno et al., 2022). C. jejuni bacteremia development correlates both with bacterial virulence and with the host ability to eliminate the bacteria (Ben-Shimol et al., 2013).
Timely and precise identification and tracing of pathogens is crucial for preventing the further spread of pathogens and controlling foodborne disease outbreaks. Whole genome sequencing (WGS) technology is increasingly popular in subtyping, due to highly precise, high-throughput capacities and abundance of genetic information available for extensive studies (Yu et al., 2020). Compared with 7-gene multilocus sequence typing (MLST), whole genome (wg)MLST is more comprehensive as it allows for examination of differences in all loci found in the reference strains used to build the allele schemes (Joseph et al., 2023). Additionally, WGS data enables the prediction of antimicrobial resistance (AMR) profiles and the identification of virulence genes in pathogens (Baker et al., 2018), which can improve the understanding of important pathogens. These advantages make WGS increasingly used around the world to facilitate surveillance, investigation, and control of foodborne diseases (Li et al., 2021).
In this study, we investigated a case of a 23-day-old infant with an invasive infection by C. jejuni, characterized the molecular epidemiology of the C. jejuni isolates in this infection, and traced the source of this infection by WGS data.
Materials and Methods
Epidemiological investigation and sample collection
On December 30, 2023, a 23-day-old infant was admitted to the local hospital (Nanchang, China), on the same day with a fever (38.9°C) of unknown cause. This infant was formula-fed with appropriate growth and development and had no relevant previous medical history, with white foam sputum at the mouth, nasal congestion, inappetence, no convulsions, no cough, no cyanosis, no diarrhea, and regular bowel movements. Upon admission, a blood sample was immediately drawn for testing. The infant’s blood was cultured in BacT/ALERT FA and FN media using an automated system (Biomerieux, France) at 37°C. Four days later, the bacterial colonies were identified as C. jejuni using by matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS, Bruker, Germany).
All of the infant’s cohabitants (the infant’s grandmother, father, and mother) were interviewed regarding their dietary, gastrointestinal illness symptoms, and poultry contact history. Additionally, anal swabs (3) from the infant’s cohabitants were collected. The infant’s feeding bottle (1), milk powder (1), kitchen knife (1), kitchen countertop (1), and chopping block (1) were also collected for detection. No leftover meals were left.
This study was approved by the Ethics Committee of the Jiangxi Provincial Center for Disease Control and Prevention (No. 202403) and performed in accordance with the Declaration of Helsinki as revised in 2013.
Strain isolation and antimicrobial susceptibility testing
All samples were added into Campylobacter enrichment broth (Zhongchuang, Qingdao, China), and then were incubated at 42°C for 24 h under microaerobic conditions (5% O2, 10% CO2, 85% N2). Two 0.45 μm pore-size membrane filters were placed on a Columbia blood agar plate (Zhongchuang, Qingdao, China) and a Karmali blood agar plate (Zhongchuang, Qingdao, China), respectively. Then, 300 μL of enrichment broth was spotted on each membrane filter. After 45 min, the filters were discarded. The plates were incubated at 42°C for 48 h under microaerobic conditions. Suspected Campylobacter colonies were identified by MALDI-TOF MS (Bruker, Germany).
Isolates were tested for antimicrobial sensitivity using the broth microdilution method. Isolates were cultured on Columbia agar at 42°C for 24 h, and then colonies were resuspended in deionized water, adjusting the turbidity to 0.5 McFarland Standard. 10 μL of the prepared bacterial suspension was added to 11 mL of Mueller-Hinton broth with lysed horse blood (Thermo Fisher Scientific, United States) and mixed well. Fifty μL of the above broth suspension was transferred to each well of the antimicrobial susceptibility test plates (Thermo Fisher Scientific, United States), including containing ciprofloxacin (CIP, 0.03–16 μg/mL), nalidixic acid (NAL, 2–64 μg/mL), streptomycin (STR, 1–8 μg/mL), gentamicin (GEN, 1–32 μg/mL), tetracycline (TET, 1–32 μg/mL), erythromycin (ERY, 0.5–64 μg/mL), ampicillin (AMP, 2–64 μg/mL), amoxicillin/clavulanic acid (AMC, 1/0.5–64/32 μg/mL), trimethoprim/sulfamethoxazole (SXT, 0.25/4.75–8/152 μg/mL), cefazolin (CFZ, 0.5–16 μg/mL), cefoxitin (CFX, 2–64 μg/mL), cefuroxime (CXM, 0.5–32 μg/mL), cefotaxime (CTX, 0.25–8 μg/mL), ceftazidime (CAZ, 1–32 μg/mL), and cefepime (CPM, 1–32 μg/mL). After inoculation, the plates were incubated at 42°C for 24 h under a microaerobic atmosphere, and then the minimum inhibitory concentration (MIC) for each antimicrobial agent was read. C. jejuni ATCC 33560 was used as control. MIC breakpoints for CIP, ERY, and TET were in accordance with the European Committee on Antimicrobial Susceptibility Testing (EUCAST, www.eucast.org) for C. jejuni. Since the MIC breakpoints for the remaining tested antimicrobials were not specified for C. jejuni by EUCAST, we used the breakpoints for Enterobacteriaceae.
WGS and bioinformatics analysis
DNA extraction was performed using a Qiagen DNeasy UltraClean Microbial Kit (Qiagen, Hilden, Germany) in accordance with the manufacturer’s instructions. Paired-end libraries (2 × 150 bp) were constructed using a Rapid Plus DNA Lib Prep Kit for Illumina (ABclonal, Wuhan, China), and sequencing was performed on an Illumina Novaseq 6000 instrument (Illumina, CA, USA). Raw reads were processed using fastp 0.23.1 for adapter trimming and quality filtering with default settings (Chen et al., 2018). Trimmed sequence reads were de novo assembled using SPAdes inbuilt in TraNet (Li et al., 2021). The presence of AMR genes and mutations associated with AMR was predicted using ResFinder 4.1 (http://genomicepidemiology.org/services/) (Bortolaia et al., 2020) with default settings. Virulence genes were determined using Virulence Factor Database (VFDB) online platform-VFanalyzer (http://www.mgc.ac.cn/cgi-bin/VFs/v5/main.cgi). Determination of allele numbers and corresponding sequence types (ST) and clonal complexes (CC) was performed by submitting the DNA sequences to the Campylobacter PubMLST website (https://pubmlst.org/campylobacter/) (Dingle et al., 2001) whole genome multilocus sequence typing (wgMLST) analysis was performed using BioNumerics 7.6, with cluster analysis based on the Categorical (differences) similarity coefficient. The Complete Linkage method was used to construct a phylogenetic tree to analyze the phylogenetic relationships between the isolates. The genomes were compared and variants detected using Snippy v4.6.0 (https://github.com/tseemann/snippy), with C. jejuni NCTC 11168 (GCF_000009085.1) as reference genome. Recombination regions were identified and filtered using Gubbins v3.4 (Croucher et al., 2015), generating a core genome alignment file. Snp-sites v2.5.1 (Page et al., 2016) was used to extract polymorphic single-nucleotide polymorphism (SNP) sites from the core genome alignment file, removing invariant positions and producing a refined sequence alignment file for phylogenetic analysis. The phylogenetic tree was constructed using IQ-TREE v2.3.6 (Kalyaanamoorthy et al., 2017) with the maximum likelihood (ML) method. The optimal model, TVMe+ASC (AIC = 55504.365, BIC = 55573.080), was selected using ModelFinder (MFP) parameter, with 1000 bootstrap replicates to assess tree robustness. The tree was rooted and visualized using iTOL (Letunic and Bork, 2024), with tree topology and branch lengths annotated to enhance clarity in illustrating the evolutionary relationships between the isolates.
Results
Epidemiological investigation and strain isolation
A 23-day-old infant was admitted to the hospital on the same day as the fever onset and was immediately subjected to blood tests. Four days later, the test results showed that the blood sample was positive for C. jejuni. Then, the infant was diagnosed with bacteremia. The infant received treatment with intravenous piperacillin sodium and tazobactam sodium (0.43 g/dose, every 8 h). After 5 days of treatment, the infant recovered and was discharged from the hospital. The epidemiological investigation indicated that the infant’s grandmother had slaughtered a live chicken at home 4 days before the onset of symptoms, and she had been responsible for brewing milk powder to feed the infant. Two C. jejuni isolates were isolated from the infant’s grandmother’s anal swab (KC3) and the feeding bottle (KC2). The remaining samples were negative for C. jejuni. All the infant’s cohabitants had no fever, no diarrhea, or other symptoms.
Typing and clustering analyses
MLST analysis identified these three C. jejuni isolates as sequence type 51 (ST51), clonal complex 443 (CC443). wgMLST clustering analysis showed isolates KC1 and KC3 were highly similar with an allele distance of 1.0, whereas KC2 was more distantly related with an allele distance of 4.0 (Fig. 1). The phylogenetic analysis based on SNPs revealed minimal evolutionary distance between isolates KC1 and KC3, with branch lengths of 1.4 × 10−5 (bootstrap: 91%). Isolate KC2 showed a short evolutionary distance from the KC1–KC3 cluster, with a combined branch length of 5.7 × 10−5 (Fig. 2). The SNP count between KC1 and KC3 was 9, between KC1 and KC2 was 12, between KC2 and KC3 was 8.
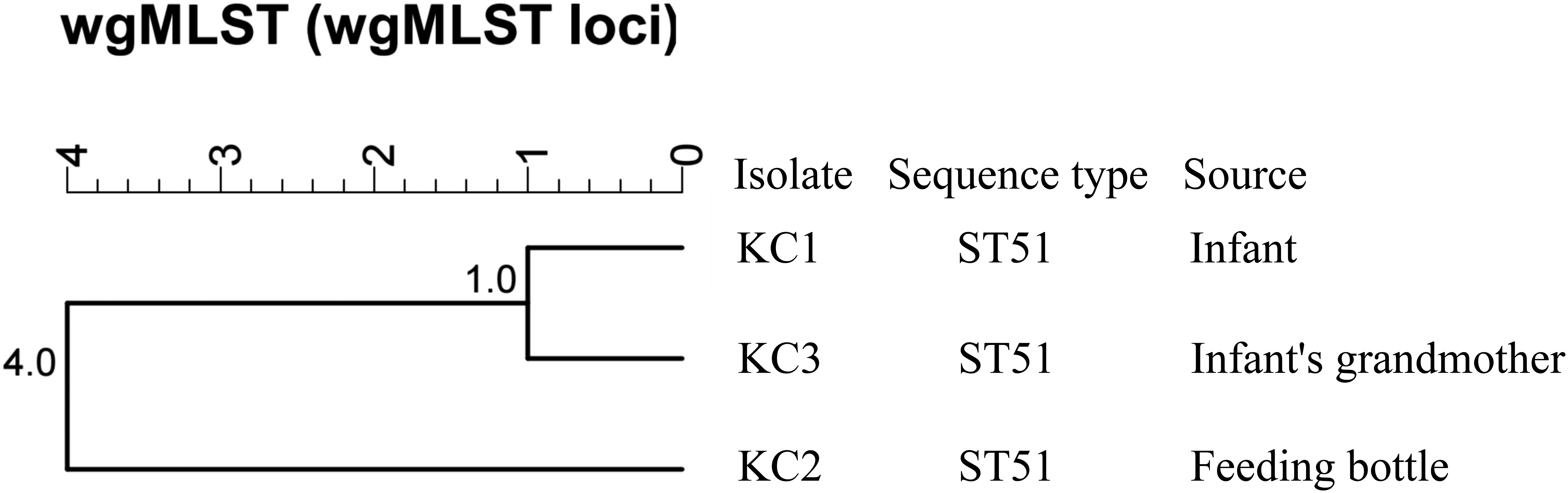
The clustering analysis of the three Campylobacter jejuni isolates, performed using wgMLST analysis with BioNumerics 7.6.
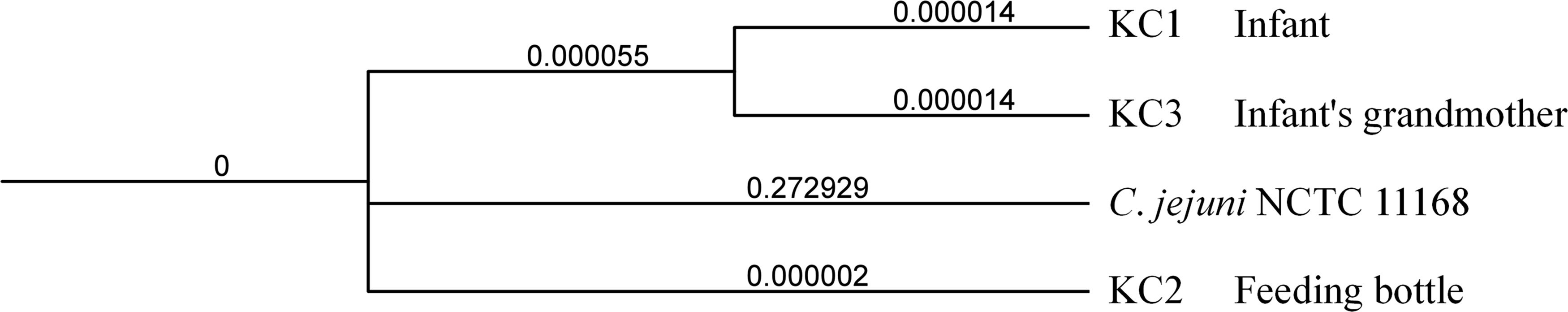
Phylogenetic tree of three Campylobacter jejuni isolates, constructed using IQ-TREE based on whole genome SNPs, generated rooted phylogenetic tree and visualized with iTOL. The branch lengths represent evolutionary distances, with numerical values indicating genetic divergence between isolates. SNP, single-nucleotide polymorphism.
Antimicrobial susceptibility and AMR genes
The three C. jejuni isolates displayed the same AMR profile (Table 1), showing resistance to fluoroquinolones (CIP, NAL) and cephalosporins (CFZ, CFX, CXM, CTX, CAZ). However, they were all susceptible to STR, GEN, TET, ERY, AMP, AMC, CPM, and SXT.
The Results of the Antimicrobial Susceptibility Testing

AMC, amoxicillin/clavulanic acid; AMP, ampicillin; CAZ, ceftazidime; CFX, cefoxitin; CFZ, cefazolin; CIP, ciprofloxacin; CPM, cefepime; CTX, cefotaxime; CXM, cefuroxime; ERY, erythromycin; GEN, gentamicin; NAL, nalidixic acid; R, resistant; STR, streptomycin; SXT, trimethoprim/sulfamethoxazole; S, sensitive; TET, tetracycline.
Prediction of AMR genes revealed that all these isolates carried the blaOXA-184 gene, conferring resistance to β-lactams. A chromosomal mutation in gyrA (T86I) was also detected (Table 2), confers resistance to CIP and NAL. No mutations in 23S rRNA or other genes (cmeR, rplV, rpsL) associated with macrolide or spectinomycin resistance were detected.
The Results of the AMR Genes Analysis

AMR, antimicrobial resistance; WGS, whole genome sequencing.
Virulence genes analysis
Sixty-nine virulence genes were found in each of three C. jejuni isolates, they shared identical virulence gene profiles. These genes were divided into five categories (Table 3), namely adherence (4 genes), motility and export apparatus (38 genes), glycosylation system (23 genes), cytolethal toxin (3 genes), and invasion (1 gene).
Functional Category of 69 Virulence Genes in C. jejuni in This Study

Discussion
This study involved a 23-day-old infant who was invasively infected by C. jejuni. To the best of our knowledge, this is the third reported case of infant bacteremia caused by C. jejuni in China to date (Wei et al., 2014; Zhao et al., 2024). These isolates were identified as ST51 (CC443), which is highly prevalent in chicken (Fiedoruk et al., 2019, Ohishi et al., 2017). Prior to the onset of symptoms, the infant’s grandmother had slaughtered a live chicken and was responsible for feeding the infant. WgMLST analysis showed that the isolates from the grandmother, the infant, and the feeding bottle had an allele distance of less than 10, indicating clonal relatedness (Zhu et al., 2021). SNPs revealed that the three C. jejuni isolates exhibited minimal SNP differences, indicating a high degree of genomic homology (Revez et al., 2014). Phylogenetic analysis further supported this observation, showing close evolutionary relationships among the isolates, suggesting that they are clonally related (Yu, H et al., 2020). The wgMLST clustering results were consistent with the SNP-based phylogenetic tree, where KC1 (infant) and KC3 (infant’s grandmother) clustered together with minimal evolutionary distance, indicating a potential direct transmission event. KC2 (feeding bottle) formed a separate branch but remained closely related to KC1 and KC3. Furthermore, in the rooted tree, KC2 appeared closer to their common ancestor than KC1 and KC3. SNPs showed that KC1 and KC2 differed by 12 SNPs, KC1 and KC3 by 9 SNPs, and KC2 and KC3 by 8 SNPs. However, these results slightly differed from the phylogenetic analysis, which may be attributed to the use of the Maximum Likelihood method and the TVMe+ASC model. Additionally, since KC2 was isolated from an environmental sample, its distinct growth conditions may have influenced its mutation profile. Moreover, SNP differences do not always correlate linearly with evolutionary distances in phylogenetic analysis. Considering the epidemiological investigation, the infant’s grandmother slaughtered a live chicken four days before the infant developed symptoms. She was likely contaminated with C. jejuni during this process and subsequently transmitted the bacteria to the infant while feeding. Although the isolate from the feeding bottle (KC2) exhibited high genetic similarity to KC1 and KC3, its SNP differences suggest that it may have acted as an indirect transmission vehicle rather than the direct source. Furthermore, we cannot exclude the possibility that both the infant and the grandmother had contact with the feeding bottle at different times, complicating the evolutionary relationship of KC2. This study highlights the importance of food safety and proper hand hygiene in household settings to reduce the risk of foodborne pathogen transmission.
In this study, the blaOXA-184 gene, which encodes resistance to β-lactams, was detected in the three isolates. However, the specific phenotype associated with this resistance to β-lactams remains unknown and requires further study. Interestingly, the three isolates showed sensitivity to AMP and AMC. This finding aligns with previous reports by Wysok et al. (2020) and Hizlisoy et al. (2023). Although Campylobacter are generally inherently resistant to many β-lactams (Wieczorek and Osek, 2013), the frequency of resistance to AMC in Campylobacter has been found to be low, specifically 0–5.8% in the studies by Post et al. (2017) and Wysok et al. (2020).
Additionally, a mutation in gyrA gene resulting in the T86I amino acid change was detected in these three isolates in this study, which confers resistance to fluoroquinolones (CIP and NAL) (Bortolaia et al., 2020), consistent with the AMR phenotype. The T86I amino acid substitution was common in quinolone-resistant C. jejuni isolates, found in 97.43% of human and 96.22% of poultry isolates, respectively (Marotta et al., 2019). High resistance to fluoroquinolones in C. jejuni was also reported worldwide: the resistance rate was >90% in Poland (Fiedoruk et al., 2019), >88.5% in China (Zhang et al., 2020), >84.2% in South Korea (Oh et al., 2017), and >45.9% Japan (Ohishi et al., 2017). Fluoroquinolones have been used to treat infections in poultry and as growth promoters over the 50 years, they may be the cause of the high resistance rate (Marotta et al., 2019). The increases in fluoroquinolone-resistant C. jejuni infections in the United States, Europe, and Asia are likely driven by the acquisition of resistant isolates from poultry (Dai et al., 2020).
In this study, 69 virulence genes were identified in each of the three C. jejuni isolates. Adhesion is a crucial step before invasion and toxin secretion (Wysok et al., 2020). The C. jejuni isolates in this study harbored genes encoding adhesion-associated proteins (cadF, porA, pebA, and jlpA) and harbored the invasion-related gene ciaB. These genes are highly prevalent in both clinical (Wysok et al., 2020; Yu et al., 2020) and animal isolates (Gharbi et al., 2022; Oh et al., 2017). The Campylobacter invasion antigen (Cia) is essential for the bacterium to invade host cells and ensure intracellular survival. CiaB has been demonstrated to contribute to diseases in vivo (Lopes et al., 2021). Another key virulence factor of C. jejuni is its ability to produce toxins. In this study, all isolates carried cdtABC genes, encoding cytolethal distending toxin (CDT), which is highly prevalent (74%–100%) in C. jejuni isolates from humans, poultry, swine, and cattle (Gharajalar et al., 2020; Lopes et al., 2021, Wysok et al., 2020). CDT, composed of CdtA, CdtB, and CdtC, induces DNA double-strand breaks, cell cycle arrest (G2/M phase), ultimately leading to senescence or apoptosis. CDT also induces interleukin-8 release, causing inflammation. Additionally, the isolates harbored 38 genes associated with motility and export apparatus, encoding the flagellar complex, which plays dual roles in host colonization and the secretion of virulent proteins (Lopes et al., 2021). Furthermore, 23 genes related to the glycosylation system were identified, falling into two categories: the N-linked protein glycosylation system (pglA
Conclusion
This study describes a rare case of C. jejuni bacteremia in a 23-day-old infant in China. WGS revealed that the C. jejuni isolates from the infant, grandmother, and the feeding bottle were clonally related, with minimal genetic divergence, confirming the close transmission link between the grandmother and the infant. The isolates exhibited resistance to fluoroquinolones and cephalosporins, with the blaOXA-184 gene and a gyrA mutation (T86I) conferring resistance. Virulence gene analysis revealed factors associated with adhesion, motility, and toxin production, which may have contributed to the infant’s invasive infection. This case highlights the importance of food safety and hygiene practices.
Authors’ Contributions
S.P.: Conceptualization; formal analysis (equal); writing—original draft; and writing—review & editing. C.L.: Project administration. H.Z.: Data curation. W.T. and J.X.: Investigation (equal). D.L.: Funding acquisition. J.L.: Formal analysis (equal).
Footnotes
Funding Information
This work was supported by Key Research and Development Program of Jiangxi Province (20243BBH81007), Jiangxi Provincial Key Laboratory of Major Epidemics Prevention and Control (2024SSY06021), Key Laboratory of Nutrition Diet and Health of Jiangxi Provincial Health Commission and Jiangxi Province High-level and High-skill Leading Personnel Training Project (2021), Science and Technology Fund Plan of Jiangxi Provincial Health Commission (202510601).
Availability of Data
The de novo sequences of strains KC1, KC2, and KC3 were submitted to GenBank of National Center for Biotechnology Information with the accession number PRJNA1174427.
Disclosure Statement
No competing financial interests exist. No personal interests to disclose.