
Abstract
The American College of Medical Genetics guidelines for microarray analysis for constitutional cytogenetic abnormalities require abnormal or ambiguous results from microarray-based comparative genomic hybridization (aCGH) analysis be confirmed by an alternative method. We employed quantitative real-time polymerase chain reaction (qPCR) technology using SYBR Green I reagents for confirmation of 93 abnormal aCGH results (50 deletions and 43 duplications) and 54 parental samples. A novel qPCR protocol using DNA sequences coding for X-linked lethal diseases in males for designing reference primers was established. Of the 81 sets of test primers used for confirmation of 93 abnormal copy number variants (CNVs) in 80 patients, 71 sets worked after the initial primer design (88%), 9 sets were redesigned once, and 1 set twice because of poor amplification. Fifty-four parental samples were tested using 33 sets of test primers to follow up 34 CNVs in 30 patients. Nineteen CNVs were confirmed as inherited, 13 were negative in both parents, and 2 were inconclusive due to a negative result in a single parent. The qPCR assessment clarified aCGH results in two cases and corrected a fluorescence in situ hybridization result in one case. Our data illustrate that qPCR methodology using SYBR Green I reagents is accurate, highly sensitive, specific, rapid, and cost-effective for verification of chromosomal imbalances detected by aCGH in the clinical setting.
Introduction
M
We chose qPCR as our primary confirmation method, but utilized banded chromosome analysis and FISH analysis for obvious translocations and common deletions. Banded chromosome analysis has limited application due to relative low resolution. Multiplex ligation-dependent probe amplification is less useful since a high percentage of abnormal aCGH findings are rare or private. FISH is widely used to confirm aCGH results and for follow-up parental studies if appropriate FISH probes are available (Baldwin et al., 2008; Bejjani and Shaffer, 2008). Advantages of FISH methods include the following: (1) FISH is in use in most clinical cytogenetic laboratories, (2) commercial FISH probes are available for common microdeletion/duplication syndromes, (3) FISH may identify mosaicism, which other methods have limited ability to resolve, and (4) FISH can provide information about chromosomal location and help clarify the mechanism for a deletion or duplication as long as the region is large enough for detection with FISH probes and metaphase chromosomes are available. Limitations of FISH methods include (1) limited availability of ready-to-use labeled FISH probes for genomic imbalances that are rare or private (Koolen et al., 2008), (2) the expense and challenge of setup and training of personnel to make and process home-brew FISH probes, (3) the expense of custom-designed FISH probes from commercial companies, (4) Bacterial artificial chromosome (BAC) clones purchased from public sources that are not specific or are annotated incorrectly (Perry et al., 2006; Redon et al., 2006), and (5) FISH probes, usually 100-150 kb, that are too large for the detection of small microdeletions or microduplications, for example, tandem duplications (Lee et al., 2007).
qPCR is a well-established method for quantifying levels of gene expression and for the detection of both constitutional and acquired genomic imbalances (Weksberg et al., 2005; Kalinova et al., 2008). TaqMan reagents have been used for confirmation of aCGH results (Mencarelli et al., 2008), but are expensive. SYBR Green I dye is a less-expensive alternative flourophore, but has not been routinely implemented. We have successfully set up and adapted qPCR using SYBR Green I dye as the primary method for the confirmation of abnormal CNVs in patients and parental follow-up studies. In the current report, a detailed description and analysis of our qPCR data are presented to highlight qPCR using SYBR Green I dye as a reliable, robust, and cost-effective method supporting aCGH testing for constitutional genomic abnormalities in the clinical setting.
Materials and Methods
Patient acquisition and DNA preparation
A total of 431 consecutive children with diverse clinical features were referred for aCGH evaluation using a high-resolution aCGH-244K platform from June 2008 to January 2009. One hundred twenty-two abnormal CNVs were identified in 102 patients. CNVs were verified by qPCR only (89), retrospective or concurrent banded chromosome analysis only (14), banded chromosome analysis plus qPCR (3), FISH analysis only (5), or FISH plus qPCR (1). Fifty-four parents (24 pairs and 6 single parents) were studied by qPCR for 34 CNVs in 30 children.
Genomic DNA samples were isolated from peripheral blood of the patients and their parents using a Gentra Pure Gene kit according to the manufacturer's instructions (Qiagen, Germantown, MD). Pooled normal human male or female genomic DNA from 6 to 10 anonymous donors of the same sex was used as reference DNA for qPCR testing (Promega, Madison, WI). The quality and quantity of DNA samples were assessed using a NanoDrop ND-1000 UV-VIS spectrophotometer (NanoDrop Technologies, Wilmington, DE) and agarose gel electrophoresis. This study was given an exempt status by the Institutional Review Board.
Identification of abnormal CNVs by aCGH testing
aCGH testing using the Agilent Human Genome Microarray Kit 244K platform was performed following the recommendation of the manufacturer (Agilent Technologies, Santa Clara, CA). Validation of the platform was described previously (Yu et al., 2009).
Primer design
Test primers targeted to DNA sequences within the aCGH-identified deleted or duplicated region were designed. Reference primers were designed using DNA sequences within genes coding for X-linked lethal diseases in males (Morleo and Franco, 2008) (Table 1). The selected DNA sequences were pasted into the University of California Santa Cruz (UCSC) genome browser for comparison with the UCSC human genome sequence version Hg18. Genomic sequences of approximately 10,000-15,000 bp without known variations were selected. Generally, the selected DNA sequences were within exonic and/or intervening regions from a functional gene. The selected DNA sequences were then pasted into IDT's PrimerQuest™ real-time PCR primer standard design software (Integrated DNA Technologies, Coralville, IA). The parameters for all designed primers were set to amplicon size of 100-200 bp with a penalty score no higher than 1.0; primer melting temperature range 60° ± 5°C; primer length of 24 bp; and primer G/C content range 50 ± 15%. Self-complementary sequences (inverse repeats), runs of more than three identical nucleotides, or more than two G and/or C bases in the last five nucleotides at the 3′ end were avoided if possible. Each set of primers was further checked for inter- and intra-molecular dimer formation. Candidate primer sequences were then tested in the UCSC In-Silico PCR program to guarantee that (1) there was a 100% match between the primer sequences and the sequence from which they were designed, (2) the forward and reverse primers were free of single-nucleotide polymorphisms, and (3) the paired primers produced expected In-Silico PCR amplification with no cross match to other genomic sequences. All primers were purchased from Integrated DNA Technologies.
qPCRs
Before utilizing qPCR for clinical cases, substantial efforts were carried out to normalize the ABI 7000 system from Applied Biosystems (Foster City, CA) (the ABI 7000 was later replaced with the ABI 7500 system), choose reagents for optimization of the method, and to standardize the methods. The Premix-Choice Kit from Epicentre Biotechnologies (Madison, WI) was selected for the reagents. Pooled normal human male or female genomic DNA from Promega was chosen as the reference DNA. Protocols for primer design and optimization were developed. The optimal qPCR conditions were standardized, and criteria for calculating results and interpretation were established.
The specificity and efficiency of all sets of test primers were first evaluated by performing optimization qPCRs with pooled normal human female genomic DNA as the reference DNA combined separately with three different qPCR buffers (buffers E, G, and I from Epicentre Biotechnologies). Each time the test primers were set for optimizing qPCRs, optimized reference primers were tested simultaneously by using the same conditions in separate reactions. Individual optimization qPCRs were performed in a 20 μL volume including 25 ng template DNA, 200 nM of each primer, and 1 × Premix-Choice with ROX reference dye in an initial denaturation of 93°C for 10 min, followed by 35 cycles of 93°C for 1 min and 61°C for 1 min, and 72°C for 1 min. Dissociation curve and agarose gel electrophoresis were used to evaluate the specificity and efficiency for each set of test primers. A satisfactory set of test primers should fit the following criteria: dissociation curve with a single narrow peak, agarose gel electrophoresis with a single sharp band of the expected size, and comparable signal intensity from amplification of reference primers. The buffer with the highest specificity and efficiency for each set of test primers was selected as the test buffer for each specific CNV assay.
When preparing assays for each CNV, there are four groups of reactions: (1) DNA plus test primers, (2) DNA plus reference primers, (3) reference DNA plus test primers, and (4) reference DNA plus reference primers. Batching of different sets of primers for different test samples sharing the same optimal qPCR buffer was performed to reduce the workload and cost. Each reaction was analyzed in triplicate for both DNA samples (test and reference samples) and both sets of primers (test and reference primers). The same optimized qPCR conditions for each set of primers were applied for the CNV testing in individual test samples. The same qPCR protocols used for confirming the abnormal CNVs in patient samples were used for parental samples. The child's DNA was run concurrently with the parental DNA as a positive control.
qPCR data normalization, calculation, and interpretation
In the current study, the comparative threshold cycle (Ct) method of relative quantitation was selected to calculate the relative DNA copy numbers in test samples. As mentioned, both test and reference primers were amplified in test and reference DNA for each abnormal CNV under investigation. The cross-reference testing was designed to correct potential variations from sampling, extracting, and processing DNA templates, PCR setup and the cycling process, as well as PCR efficiency. Results were expressed in terms of the Ct value at which the fluorescence intensity for the SYBR green I dye with ROX as the passive reference dye exceeds the detection threshold. The results were recorded using the ABI 7500 detection system and associated SDS software version 2.1 (Applied Biosystems), which allows export to a tab-delimited text file format. Further calculations were performed using Microsoft Excel. With normalized Ct data, results from each test sample were compared to the results from the corresponding reference sample. Thus, any normalized differences of Ct values between test and reference samples reflect the differences of copy number of test locus in test and reference samples. The formula used for the copy number calculation is 2 × 2ΔΔCt (the actual copy numbers of test gene in test samples), where ΔΔCt = ΔCt (reference sample) − ΔCt (test sample). ΔCt represents the difference between the Ct of a test gene and the Ct of the selected reference gene. Cut-off values for duplication and deletion were set arbitrarily with ΔΔCt = 0.585 ± 0.20 (0.585 × 35%) for a duplication, and ΔΔCt = − 1 ± 0.35 (1 × 35%) for a deletion (e.g., based on the formula, the accepted actual copy number for a deletion should be 1 ± 0.35 and for a duplication it should be 3 ± 0.35). Any qPCR test result outliers were repeated until consistent results were achieved.
Results
Evaluation of experimental procedures and validity of designed primers
All DNA samples had a 260/280 ratio greater than 1.6 checked by spectrophotometer, and none showed DNA degradation in agarose gel electrophoresis (data not shown). Primer designs and ordering were straight forward and robust. Ideal design for primers is prerequisite for successful qPCR experiments. In the current study, a total of 81 primer pairs for 93 CNVs (50 deletion and 43 duplication) in 80 patients were required for aCGH result confirmation and parental follow-up studies (Table 1). The size of the qPCR amplicons ranged from 100 to 199 bp. All sets of test primers were first evaluated by performing optimization qPCRs. Consequently, 71 of the 81 test primer sets worked well after the initial design and optimization. Of the remaining 10 test primer sets, 9 sets were redesigned and optimized in a second round and 1 set in a third round because of no PCR amplification or spurious products. The coefficient of variations for the Ct values of the triplicates within each reaction was lower than 5%, thus showing great accuracy of our qPCR system. The repeat rates for our qPCR testing were 61% using two-copy reference primers and 34% using single-copy reference primers.
qPCR result comparisons between single-copy reference primers and two-copy reference primers
Of 81 sets of different test primers for CNV verification, 50 were tested using one of the two-copy reference primers (NEGR1, PRKAB2, TLR4, and WWP1), and 31 were tested using one of the single-copy reference primers (CCHS and OFD1 genes). These genes are on the X chromosome and male DNA was used as a reference; thus, there is only a single copy present. When these two single-copy reference primers were used, our qPCR repeat rate was reduced from 61% to 34%.
Confirmation of abnormal aCGH results in patients and parental follow-up studies
Ninety-three CNVs (50 deletions and 43 duplications) in 80 patients were confirmed by qPCR methods using 81 sets of test primers. The aCGH results were concordant with the qPCR results for 91 CNVs. The aCGH results in two cases were clarified with the qPCR results. In one case, the qPCR result from initial and repeat tests showed four copies of the test gene (ARHGEF9). Review of the aCGH result showed a triplication instead of a duplication (Fig. 1a). In a second case, the aCGH result was initially reported as a single-copy deletion of 15q13.33. However, the qPCR result showed consistent lack of amplification in the DNA sample using a set of test primers for CHRNA7, a gene within the deleted interval (Fig. 1d). The aCGH image corroborated this as a homozygous 1.5 Mb deletion, rather than a hemizygous deletion.
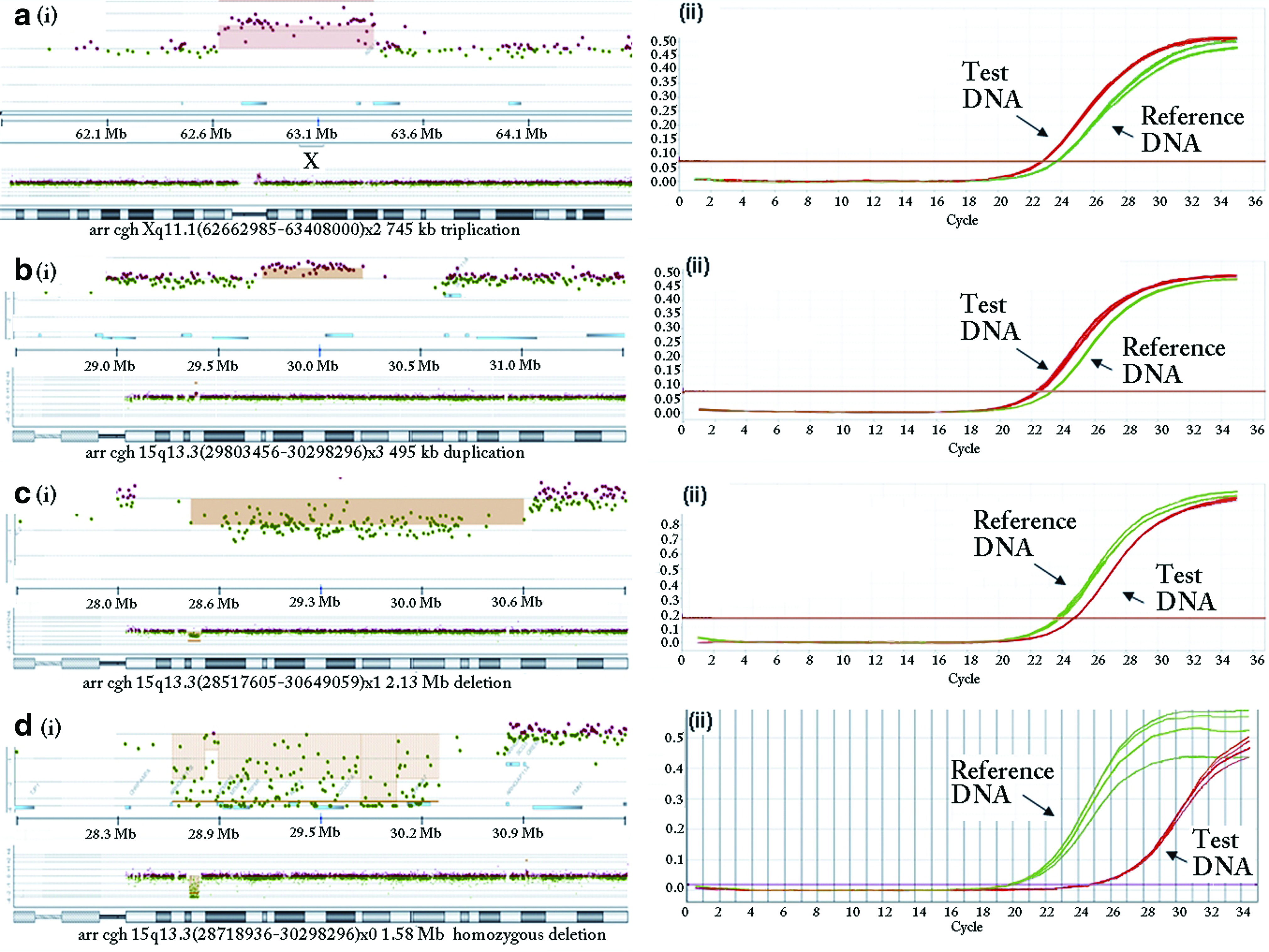
(
In a third case, qPCR confirmed the aCGH finding of a 600 kb terminal deletion of chromosome 17q when a commercially available 17q subtelomere FISH probe located within the deleted region indicated a normal signal pattern with no deletion. qPCR using primers targeting the ZNF750 gene within the deleted interval confirmed a single-copy deletion (Fig. 2a-c).

(
A fourth case demonstrated the inability of aCGH and qPCR to determine rearrangement status. Figures 3a and 3b show a single copy of 14q32.31q32.33 and three copies of 16q24.3, respectively. aCGH and qPCR indicate the imbalance, but cannot detect a balanced translocation that may be carried by a parent. Metaphase FISH analysis revealed that the single copy of 14q32.33 and three copies of 16q24.3 were the results of an unbalanced translocation with one 16q24.3 signal on a derivative chromosome 14 (Fig. 3c, d). In this case a parent had a balanced translocation that would have gone undetected if tested only by qPCR.

(
qPCR was used to test 54 parental DNA samples (24 couples and 6 single parents) using 33 sets of test primers to determine the inheritance pattern for 34 CNVs in 30 children. Confirmation was successful for 33 of the 34 CNVs. One case showed a 2.0 Mb deletion of chromosome 12q24.13q24.21 by aCGH. Using a set of two-copy reference primers as an internal control, the deletion was confirmed in the patient without difficulty using a set of test primers targeted to the TBX5 gene. However, the maternal DNA showed ∼1.5 copies of the TBX1 gene in two different runs. Mosaicism for the deletion in the mother was initially suspected. aCGH testing of the maternal DNA using the aCGH-244K chip showed two copies of the TBX5 gene. Recheck of the maternal DNA sample using the same set of test primers for the TBX5 gene but with a set of single-copy reference primers found two copies of the TBX5 gene. This is a result of improved resolution to copy number change when standardizing against single-copy reference genes.
Nineteen of the 34 CNVs with available parental samples were inherited, 13 were negative in both parents, and 2 were inconclusive due to a negative result from a single parent.
Estimation of qPCR turn-around time and experimental cost
Turn-around time is generally 2 weeks for qPCR confirmation if a new pair of primers must be designed. The design takes approximately 45 to 60 minutes; ordering and receipt, 3-4 business days. After receipt, it takes about 1 day for the primers to be registered in the database, hydrated for stock and working concentrations, and optimized. If the primers satisfy the criteria for specificity and amplification efficiency, the primers are used to confirm a CNV in the test sample. It takes 1 day from qPCR preparation to data analysis. Different sets of test primers for different DNA samples with the same optimal qPCR buffer are run in parallel to reduce workload and cost. The cost of each set of primers with potential application for hundreds of reactions is ∼$26. The qPCR reagent cost for confirmation of each CNV is ∼$15-20, including repeated tests using 20 μL for each reaction. In total, the reagent costs for qPCR was approximately $50 per CNV confirmation. This cost will decline as our library of primers expands.
Discussion
We have systematically explored the clinical application of qPCR as a method for confirmation of pathogenic CNVs or CNVs of uncertain clinical significance identified with the aCGH-244K platform. The qPCR protocol that we established was used to verify all 93 abnormal CNVs using 81 sets of test primers. These primer pairs were subsequently used for 54 parental follow-up studies. Of the 81 test primer pairs, 71 sets (88%) worked optimally with the initial design. Nine sets were redesigned once and one set needed a second redesign for optimization. We introduced the use of DNA sequences in genes coding for X-linked lethal diseases in males for the design of reference primers for the internal control to accurately calculate the DNA copies of CNVs in test samples. Using two sets of a single-copy reference gene, the qPCR repeat rate was reduced to 34%. Our results demonstrate that qPCR using SYBR Green I reagents is a reliable, robust, and cost-effective method supporting aCGH testing for constitutional cytogenetic abnormalities in the clinical setting. Additionally, the qPCR results reflect the accuracy and reliability of the aCGH-244K platform for detection of CNVs.
The discrimination of a single-copy deletion or duplication in a test sample from two copies in a reference sample approaches the limits of resolution of the qPCR methodology using SYBR Green dye. Steps that are critical for reliable results include high-quality test and reference DNA, selection of optimal reference DNA primers, specific and efficient amplification of test and reference primers, calibrated qPCR equipment, and skillful setup of the qPCR.
We chose the “Relative Standard” algorithm as the quantitation method for the determination of the copy number(s) of a CNV in a test sample as it results in fewer total reactions. We found that 25 ng of genomic DNA from both test and reference samples as qPCR templates provided the optimal range of 22-25 cycles of amplification for each set of test primers.
To reduce individual variation in human genomic regions, we chose pooled human normal genomic DNA from Promega as the reference DNA. The mixed genomic DNA should contain two copies (or close to two copies) of DNA sequences across the human genome.
Initially, we used four sets of primers designed from genes on autosomal chromosomes (NEGR1, PRKAB2, TLR4, and WWP1) for reference. Although these reference primers and the test primers showed satisfactory specificity and amplification efficiency, our qPCR repeat rate reached 61% as a result of reduced resolution when the reference had two copies of the target sequence. Introduction of single-copy reference primers from the DNA sequences for X-linked lethal diseases in males (CCHS and OFD1) reduced the qPCR repeat rate to 34% due to improved sensitivity in relationship to the reference gene. The rationale for designing reference primers from X-linked lethal genes included (1) the reduced likelihood of variability in copy number due to their important function and (2) the calculation window (resolution) for a duplicated CNV is expanded leading to increased accuracy. For example, if a test CNV is a duplicated region on one of the autosomal chromosomes, the ΔCt, which reflects the difference of threshold cycles between amplification for the test gene and reference gene, should be log23/1 = 1.585 (using single-copy reference primer), rather than log23/2 = 0.585 (using two-copy reference primer). The accuracy of calculation for a duplication or deletion is increased using this method.
As the high-resolution aCGH methodology becomes more commonly used in the clinical setting, smaller microdeletions, microduplications, and CNVs of uncertain significance will be detected and will need to be confirmed. While FISH methods are used almost exclusively in some laboratories now for confirmation and parental follow-up, the qPCR advantages of increased accuracy and greater flexibility will make this method increasingly useful.
Our laboratory routinely performs FISH that was utilized to confirm five CNVs. In one case, aCGH showed a 600 kb terminal deletion of chromosome 17q. A commercial 17q subtelomere FISH probe located within the deleted region showed a normal signal pattern in interphase and metaphase analysis. qPCR with primers targeting the ZNF750 gene within the deleted region confirmed a single-copy deletion (Fig. 2a-c). Subtelomere FISH analysis of 11,688 individuals with developmental disabilities found abnormalities in 2.5% (Ravnan et al., 2006). No cases were observed with a 17q subtelomeric deletion. This may reflect ineffectiveness of a commonly used commercial probe. Some FISH probes were recently shown to be inaccurate, illustrating the risk of ambiguous or incorrect results with a rare or private CNV (Kidd et al., 2008). Other examples of FISH probe inaccuracy include redesign of BAC-clone aCGH platforms to eliminate BAC probes with incorrect annotation (Shaffer and Bejjani, 2004), and an update to the Database of Genomic Variants to separate CNVs identified by BAC clone probes from non-BAC clone probes (http://projects.tcag.ca/variation/project.html). The availability of validated FISH probes useful for confirmation of small deletions and duplications is limited. With the detection of rare or private CNVs by high-resolution genome-wide aCGH, it will become increasingly difficult and expensive to confirm these with FISH. Due to the diversity of the 93 CNVs detected by our high-resolution array platform, not all could have been verified by FISH. The primers we have acquired are part of our primer library and can be used for future samples. Another advantage of qPCR over the FISH method is that the DNA used for aCGH testing can be used for confirmation. An additional blood sample is needed for FISH confirmation.
qPCR does not elucidate the mechanism behind genomic imbalances. Therefore, the cytogeneticist must use all information to determine the optimal confirmation method. An example is a case with an aCGH finding of a terminal deletion of 14q and a terminal duplication of 16q (Fig. 3a-d). FISH analysis showed an unbalanced translocation between 14q and 16q in metaphase nuclei. It must be considered that the derivative chromosome 14 was inherited from a parent with a balanced translocation. If only qPCR is used to evaluate parental samples, a false-negative result would be obtained from a parent with a balanced translocation. FISH analysis can be used for aCGH confirmation and parental follow-up studies when possible to elucidate the mechanism. Detection of mosaicism is poorly assessed with qPCR. If mosaicism is suspected, FISH should be employed if suitable probes are available.
Our results demonstrate that qPCR utilizing SYBR Green I dye is an accurate, highly sensitive, specific, rapid, and cost-effective method for verification of chromosomal imbalances detected by aCGH assays. This method will be increasingly useful as utilization of high-resolution aCGH platforms expand in the clinical service and research areas and smaller copy number variations need to be investigated.
Footnotes
Acknowledgment
The authors thank Dr. David Zwick for his support.
Disclosure Statement
No competing financial interests exist.