
Abstract
Chronic granulomatous disease (CGD) is a rare primary immunodeficiency affecting the innate immune system. Even if functional tests address the diagnosis of CGD, the identification of a molecular defect is essential for counselling family members at risk for being CGD carriers and for prenatal diagnosis. The X-linked form occurs in 65% of CGD patients. It is due to mutations in the CYBB gene, up to 12% of which are caused by large deletions. CGD carriers are usually healthy, and molecular analysis is essential to reveal their carrier status. The aim of this study was to apply a gene dosage approach, using SYBR green quantitative real-time polymerase chain reaction (RT-PCR), to quantify the genomic copy number in carriers and noncarriers of gross deletions covering the region of the CYBB gene. We studied the expression of two different amplification products of the CYBB gene, and the results confirmed a highly reduced expression of the gene in the carrier samples. The results were confirmed by linkage analysis and fluorescence in situ hybridization. Quantitative real-time PCR is fast and simple to perform, and we propose it as a new routine diagnostic approach to detect CGD carriers of deletions covering the region spanning the CYBB gene.
Introduction
C
The DHR test is a sensitive diagnostic screening for identification of female carriers of X-CGD (Bohler et al., 1986; Crockard et al., 1997). DHR oxidation does not strictly correlate with NADPH oxidase activity. An uninformative pattern of oxidase activity could be revealed both in a small percentage of X-CGD patients (Mauch et al., 2007) and in cases of skewed X-chromosome inactivation (Van Pelt et al., 1996; Köker et al., 2006). The diagnosis of female carriers becomes difficult in such cases. Although sequence analysis is indispensable to identify the presence of genetic mutations in CGD patients and their relatives, it is inadequate to reveal large deletion mutations (Joncourt et al., 2004). Two approaches have been used to determine large deletions: fluorescence in situ hybridization (FISH) and linkage analysis. FISH is a well-established method to identify chromosomal microdeletion syndromes and carriers of the X-linked diseases, as already described for X-CGD (Simon et al., 2005). Polymorphic markers included in the deleted region are usually employed in the linkage analysis, where their heterozygosity or hemizygosity can identify if one of the two X chromosomes carries the deletion, but the markers are not always informative (Gorlin, 1998). Herein we describe a new quantitative real-time polymerase chain reaction (RT-PCR) method, using the unspecific dsDNA dye SYBR green, to detect carriers of deletion covering the region spanning the whole CYBB gene (Kannengiesser et al., 2008). We validated the results through linkage analysis and FISH.
Materials and Methods
Patients
Two unrelated male patients from the Italian registry of chronic granulomatous disease, which is part of the Italian primary immunodeficiency network (IPINET), were tested for mutations in the CYBB gene. They were diagnosed as having CGD on the basis of their clinical history and the inability of their phagocytes to generate reactive oxygen species by conventional granulocyte functional tests as previously described (Martire et al., 2008). Molecular analysis of CYBB gene revealed that genomic DNA from both patients did not amplify any of the 13 exons of the CYBB gene. We therefore hypothesized the presence of a deletion covering the region spanning the whole gene, and the analysis of patients and their mothers by FISH confirmed this. The results have been published and are available upon request (Di Matteo et al., 2009).
In the first family (Fig. 1A) the mutation affects the proband, his mother, and his sister, while in the second family (Fig. 1B) only the proband has deletion in the CYBB gene.
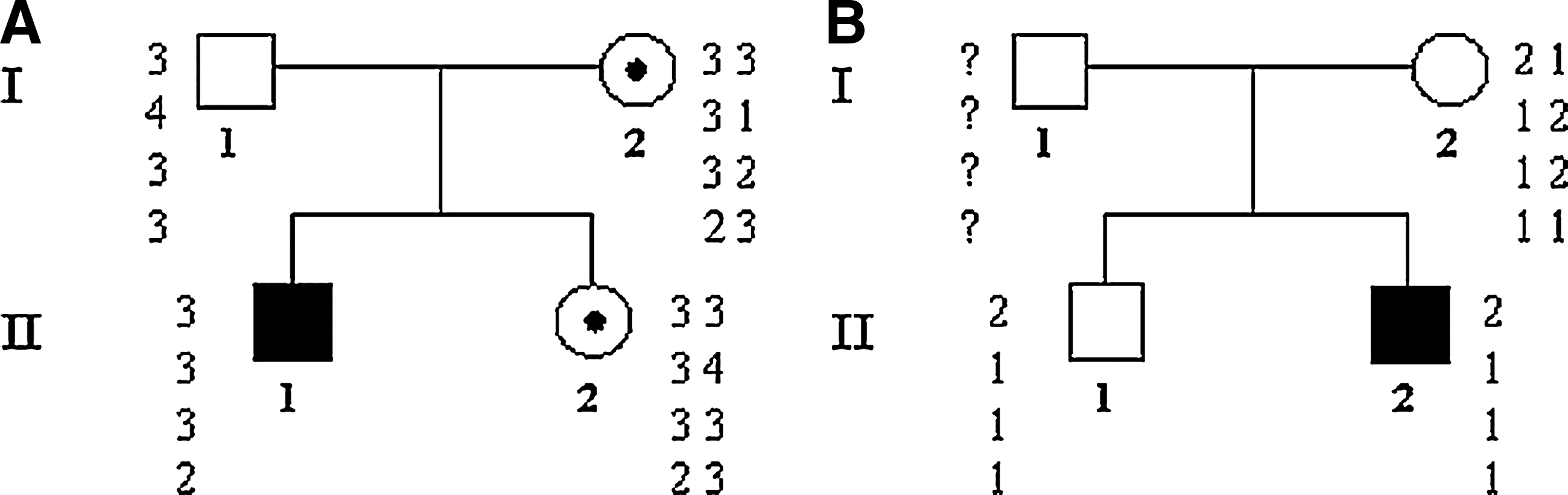
Pedigrees and haplotypes of two unrelated families. (
Informed consent was obtained from the parents of each child, and all procedures used were in agreement with the guidelines of the Helsinki Declaration on Human Experimentation.
All studies were performed at the Pediatric Immunology and Biotechnology Laboratory, Tor Vergata University of Rome. FISH studies were performed at Lombardi Comprehensive Cancer Center, Georgetown University Medical Center, Washington, DC.
Sample preparation
Cell lines
Epstein-barr virus (EBV)-transformed lymphoblastoid cell lines were obtained by incubating about 5 × 106 peripheral blood mononuclear cells (PBMC) from patients, relatives, and healthy donors with 2 mL of supernatant from the EBV-secreting cell line B95-8 for 1 hour at 37°C. Infected cells were then washed and cultured at 1 × 106 cells/mL in RPMI 1640 (Sigma-Aldrich Corp. St. Louis, MO) containing 25 mM Hepes, 10% fetal bovine serum (Sigma-Aldrich Corp.), 2 mM L-glutamine (Sigma-Aldrich Corp.), and 1 μg/mL cyclosporin A (Sandimmun, Novartis Farms S.p.A., Origgio-Varese, Italy) in 24-well culture plates. Clumps of transformed cells were usually observed at the end of the first week, and stable EBV lymphoblastoid cell lines were established by 3-4 weeks.
RNA extraction and RT-PCR
Total RNA was isolated from EBV-transformed B lymphocytes of CGD patients, relatives, and healthy donors by using TRIzol reagent procedure (Invitrogen Life Technologies, Milan, Italy). Isolated total RNA was analyzed by spectrophotometer (OD 260), and 500 ng was used for cDNA synthesis carried out with random primers and SuperScriptTM III First-Strand Synthesis SuperMix (Invitrogen Life Technologies) in a total volume of 20 μL. Reverse transcription was done in accordance with the manufacturer's instructions.
Genetic study
RT-PCR
The expression study was performed in a 96-well optical reaction plate using an ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA) with Real Master Mix (Eppendorf AG, Hamburg, Germany). PCR conditions were in accordance with the manufacturer's instructions. PCR were set up in a total volume of 25 μL containing 2.5x Real Master Mix/20x SYBR solution, 200 nM one primer pair, and 5 μL of cDNA adjusted to 10 ng. We chose GNB2L1 gene as the genomic reference and used two different amplification products of CYBB gene to optimize this diagnostic protocol. The following primers were designed using IDT SciTools Primer Quest SM software (www.idtdna.com) to generate amplicons between 300 and 400 bp and with melting temperature 57°C: GNB2L1ex3 forward 5′-GAG TGT GGC CTT CTC CTC TG-3′, GNB2L1ex5 reverse 5′-GCT TGC AGT TAG CCA GGT TC-3′; CYBBex1 forward 5′-ACT GGG CTG TGA ATG AGG G-3′, CYBBex3 reverse 5′-CGA CAG ACT GGC AAG AGA ATC-3′; CYBBex7 forward 5′-GGA ATG CCC AAT CCC TCA G-3′, CYBBex8 reverse 5′-CCT TCT GTT GAG ATC GCC A-3′. The PCR program was initiated at 95°C for 4 min, followed by 40 cycles of 30 seconds at 94°C, 30 seconds at 57°C, and 30 seconds at 72°C.
In each assay, two healthy controls (female and male), patients, relatives, and no-template control were run in triplicate, and each experiment was repeated three times. The standard curves for both primer pairs resulting from the amplification of 2, 4, 8, 16, and 36 ng of control cDNA was prepared. GNB2L1 standard curves: slope = −3.077, R2 = 0.992; CYBB1F/3R stan-dard curves: slope = −3.141, R2 = 0.956; CYBB7F/8R standard curves: slope = −3.041, R2 = 0.972. Absolute quantification of target amplicon in the patient was performed by interpolating the threshold cycle number (Ct) against the corresponding standard curve. Using this method we assumed that the rate changes of Ct are identical for GNB2L1 and CYBB genes. The control was normalized to value 1.
Microsatellite genotyping
Genomic DNA was extracted from peripheral blood lymphocytes by standard procedures (QIAamp DNA blood kit from QIAGEN GmbH, Hilden, Germany). Four short tandem repeats (STRs) markers—DXS8090 and DXS1068 localized upstream of CYBB gene, and DXS8102 and DXS8015 localized downstream of CYBB gene—were selected for this study and typed in both families. The markers were selected from the UCSC genome browser, available at http://genome.ucsc.edu. Heterozygosity value, exact position, and range of allelic size are reported in Table 1. Amplification of the markers was performed by True Allele PCR Premix (Applied Biosystems). PCR products were prepared in accordance with the manufacturer's instructions and ran for 24 min on an ABI 3130 XL automated sequencer (Applied Biosystems) in the presence of LIZ-500 fluorescent size standard (Applied Biosystems) and formamide. GENEMAPPER software version 3.7 (Applied Biosystems) was used for data collection and determination of allele size.
Results
The first analysis was performed in both families using GNB2L1 gene as genomic reference and CYBB1F/3R amplification products to discriminate deletions covering the region spanning the whole CYBB gene (Fig. 2A). In each assay, the control samples were separated into male and female controls (ctr F and ctr M) to show that the expression of GNB2L1 and CYBB genes were not affected by gender. Each bar represents 2−ΔΔCt value obtained for each sample analyzed in triplicate in three different experiments. In particular, II.1A and II.2B are the male probands showed in Figure 1, with the deletion of the CYBB gene. I.2A and II.2A are the mother and the sister of proband A, respectively, who show a carrier pattern with heterozygous deletion of the same region and a highly decreased level of mRNA expression. I.2B is the mother and II.1B is the healthy brother of proband B showing a normal pattern.
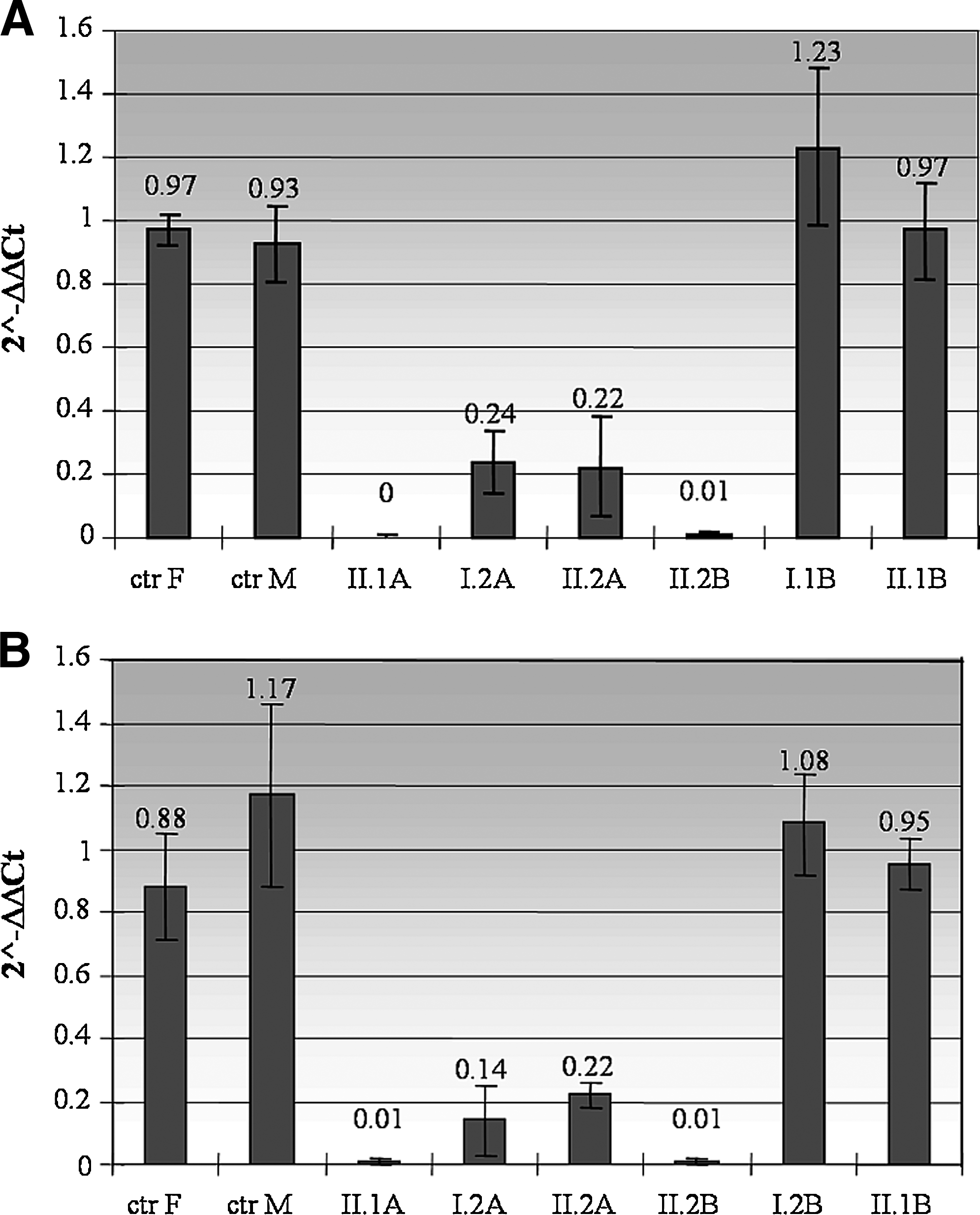
Real-time PCR (RT-PCR) results calculated by ΔCt method. Each bar represents values obtained from the analysis of all samples in three experiments, normalized to a normal control DNA. II.1A and II.2B are the male probands with the deletion of CYBB gene; I.2A and II.2A, mother and sister of proband II.1A, show a carrier pattern with heterozygous deletion of the same region. I.2B and II.1B, mother and brother of proband II.2B, show a normal pattern. (
All 2−ΔΔCt values were collected to calculate the mean and the final standard deviation for each analyzed sample. To validate the results of the first analysis, a second analysis was performed in both families using a different amplification product, CYBB7F/8R. The data obtained confirmed the previous results (Fig. 2B).
The specificity of the amplification product was confirmed by direct sequencing. The dissociation stage of RT-PCR showed the absence of primer dimers formation (data not shown). Finally, the data in Table 2 show the mean value of each group of samples (carriers, positive control with gene deletion, and male and female negative controls) and the variation range of results. Together, these results confirm that a carrier pattern with heterozygous deletion of CYBB gene in females can be identified by quantitative RT-PCR.
SD, standard deviation.
Microsatellite analysis and FISH
We made haplotypes for each family using the four STR markers (DXS8090 and DXS1068 both localized upstream, DXS8102 and DXS8015 localized downstream of the gene; Table 1). As shown in Figure 1A, in the first family the affected haplotype is present in the proband, his mother, and his sister (II.1, I.2, and II.2), while in the second family (Fig. 1B) the same haplotype segregates from the mother to the proband and his healthy brother. In the latter family, the microsatellite analysis could indicate a de novo deletion of the proband II.2B although a germ-line mosaicism in his mother cannot be excluded.
Analysis by FISH, performed as already described by Simon et al., 2005 confirmed the presence of one copy of the CYBB gene in the mother and sister of proband II.1A and two copies of the gene in the mother of proband II.2B (showed in Di Matteo et al., 2009).
Discussion
CGD is caused by mutations in any one of four genes that encode the subunits of the phagocyte NADPH oxidase. The X-linked form that occurs in 65% of CGD patients is due to mutations in the CYBB gene, up to 12% of which are caused by deletions of the gene.
The clinical phenotype of X-CGD patients is characterized by severe bacterial and fungal infections, predominantly of the lymph nodes, subcutaneous tissue, lungs, liver, and bones. The disease is normally diagnosed in young children, and the identification of CGD carriers usually does not occur until a symptomatic proband is diagnosed. Females in CGD families are at risk of being carriers, and their detection is of great importance for genetic counselling, prenatal diagnosis, and for their life expectations. Indeed, few cases of symptomatic female carriers due to a skewed X chromosome inactivation have been reported (Anderson-Cohen et al., 2003; Köker et al., 2006). Moreover, a high incidence of cutaneous and other lupus-like symptoms in carriers of X-CGD have been described (Cale et al., 2007). Female carriers are usually detected by biochemical analyses such as NBT and DHR 123 assay. However, these determinations can be somewhat subjective, and the ability to accurately identify carriers might vary from laboratory to laboratory. This is particularly true in those rare cases (about 2% of all X-CGD cases; 0.2% if we consider deletion-type mutations) where the random process of X chromosome inactivation in X-CGD carriers may determine a near-normal or a near-pathological pattern in these tests. Also, a fresh blood sample is not always available for the DHR analysis, particularly when the patients' and their relatives' samples come from areas distant from the place where molecular diagnosis is performed, which happens in the majority of cases. Therefore, once the family-specific mutation is known, it is more reliable to perform carrier detection at the DNA level. Molecular tests are usually inadequate to reveal large deletion mutations because of the sole amplification of the wild-type allele.
The aim of this study was to develop a simple and reliable assay for direct detection of X-linked CGD carriers of large deletions covering the region spanning the whole CYBB gene. Both patients presented in this work were characterized by such a deletion, and the carrier status of their mothers had been previously identified by FISH (Di Matteo et al., 2009). We report here a rapid quantitative RT-PCR approach using the unspecific dsDNA dye SYBR green to quantify the genomic copy number of the CYBB gene. In the first family (Fig. 1A) the deletion is present in the proband, while his mother and sister are carriers; in the second family (Fig. 1B) only the proband is mutated. We used two different amplification products of CYBB gene to optimize this diagnostic protocol. The specificity of the amplification products was established by direct sequencing. Moreover, the data were validated by standard curves repeated in each experiment and confirmed in both healthy control and carrier samples. The results obtained by RT-PCR were validated by microsatellite analysis. The pedigree analysis was optimized using four microsatellites with high variability among the population and located in CYBB gene (Xp21.1). In the first family (Fig. 1A) the affected allele is present in the proband's sister and mother; in the second family (Fig. 1B) the same allele segregates from mother to both siblings, but only the proband is affected revealing a case of de novo mutation.
The RT absolute quantification PCR, like the classical RT semi-quantitative PCR, requires stringent quality control, standardization of the sample acquisition, and processing. However, RT-PCR is significantly less variable than any conventional quantification PCR procedure. Moreover, the great advantage of the proposed method is that it requires the same pairs of primers commonly used in routine molecular diagnostics of CGD. Likewise, in principle, any CYBB deletion, covered by a specific primer pair, could be used in this diagnostic protocol to establish the carrier status.
RT-PCR is a new rapid method to identify large deletions. This technique is simple to perform, and we propose it as a routine diagnostic technique to detect CGD carriers of deletions.
Footnotes
Disclosure Statement
No competing financial interests exist.