
Abstract
X-linked Charcot-Marie-Tooth (CMTX) disease is a hereditary motor and sensory neuropathy caused by mutations in the gap junction beta 1 gene (GJB1 codes for connexin 32). In this study we report six novel mutations p.Met1Arg, p.Leu9Phe, p.Ser17Tyr, p.Val63Phe, p.Val170Ile, and p.Leu212Phe in GJB1 and their phenotypic expression. These mutations affect both intracellular and extracellular parts of the GJB1 protein. The screened patients had previously excluded the duplication/deletion on 17p11.2 and the male-to-male transfer in the pedigree. Except p.Val170Ile, all reported mutations segregated with the CMT phenotype in the families and caused CMTX1 neuropathy. Mutations were not found in 200 control DNA samples. Additionally, we performed in silico analysis of the novel mutations with the program PANTHER. The PANTHER scored five mutations, all but p.Val170Ile, as likely deleterious and supported the pathogenicity of the found mutations. These results provided evidence that these five mutations are causative for CMTX1.
Introduction
C
The X-linked CMT (CMTX1) is the second most frequent form of CMT (Ionasescu, 1995; Nelis et al., 1996; Casasnovas et al., 2006) with mutations in the gap junction beta 1 (GJB1) gene coding for connexin 32. Nerve conduction velocities in most of the affected males with GJB1 mutations are intermediate and range between 25 and 40 m/s in upper limb motor nerves (Nicholson and Nash, 1993). In women, CMTX1 is typically milder, and the women are affected later than men with less severe atrophies and deformities. Further MNCV in affected women are close to normal values (Hahn et al., 1990; Nicholson and Nash, 1993).
GJB1 is probably the second most commonly diagnostically tested gene in patients with CMT. GJB1 is a small gene that encodes a gap junction protein beta 1 (connexin 32), which forms the intracellular channels and facilitates the transfer of ions and small molecules in the paranodal myelin loops and Schmidt-Lanterman incisures of myelinating Schwann cells in the peripheral nervous system (Bergoffen et al., 1993). GJB1 is highly conserved among species and is expressed in many tissues; however, the peripheral neuropathy is the main clinical manifestation of GJB1 mutations (Bone et al., 1997). Several reports published associations of some GJB1 mutations with subclinical central nervous system involvement (Nicholson and Corbett, 1996; Nicholson et al., 1998; Stojkovic et al., 1999; Matsuyama et al., 2001; Seeman et al., 2001).
More than 290 different mutations in the GJB1 have been reported including missense (the most frequent type), nonsense mutations, deletions, insertions, and frameshifts which affect the coding region as well as the untranslated region of GJB1 (www.molgen.ua.ac.be/CMTMutations/). Mutation affecting the initiation codon has not yet been observed and its effect on patients' phenotype is unknown.
We report six novel mutations in the GJB1 gene, including the first mutation affecting the initiation codon, and describe their phenotypic expression in affected families. We also report a very rare case of a de novo mutation in GJB1 and provide evidence that p.Val170Ile is a rare polymorphism.
Materials and Methods
Czech patients with CMT were selected for GJB1 gene sequencing as part of routine diagnostic DNA testing. The chosen patients fulfilled the general phenotypic and electrophysiological criteria of CMT. There was no male-to-male transmission in the pedigree. Genomic DNA was extracted from blood samples or saliva using standard protocols. The CMT1A duplication/hereditary neuropathy with liability to pressure palsies (HNPP) deletion was previously excluded in all patients using the set of 17 microsatellite markers (Seeman et al., 2000). All patients have signed informed consent with the analysis of hereditary neuropathies related genes.
The coding region of GJB1 gene was amplified in one polymerase chain reaction using primers cx32 0F and cx32 3R. Direct sequencing of exon 2 was performed in two fragments using primers cx32 0F and cx32 2F (sequences are available upon request) and analyzed on an ABI3100 Avant Genetic Analyzer (Applied Biosystems, Foster City, CA). The sequence data were analyzed using the ABI DNA Sequencing Analysis software 3.7.
When the presence of mutation was observed, samples from additional family members were analyzed to evaluate the segregation of the mutation with the disease.
Two hundred ethnically matched DNA samples were analyzed by sequencing, and in addition, 165 additional DNA controls were analyzed using the SNaPshot method (ABI, Foster City, CA) focusing on p.Val63Phe and p.Leu9Phe mutations.
We used the sequence alignment of GJB1 gene from www.ncbi.nlm.nih.gov. The probability of a deleterious effect on protein function was made with in silico analysis using the program PANTHER (www.pantherdb.org) (Thomas et al., 2006). We used the protein reference sequence MIM NP_001091111.
Results
Six novel GJB1 mutations are summarized in Table 1. All six new mutations are missense mutations present in six different families. These mutations were not found in 200 DNA controls. The subPSEC values below −3 from in silico analysis of program PANTHER refer to the disease-causing mutations (Thomas et al., 2006). The pdeleterious values, the probability that the mutation will cause a deleterious effect on protein function, are high in five of the novel mutations, namely, p.Met1Arg, p.Leu9Phe, p.Ser17Tyr, p.Val63Phe, and p.Leu212Phe. These five mutations (except the p.Val170Ile) were not detected in healthy relatives.
In the column “Family history” the number of relatives with the same mutation is given within parentheses.
Clinical characteristics of patients carrying the reported GJB1 mutations are summarized in Table 2. Patients mostly showed the typical CMT phenotype with distal muscle weakness and atrophy more pronounced in the legs, decreased or absent reflexes, sensory loss, and feet deformities. In summary, men are more severely and earlier affected than women carrying the same mutations and showed more pronounced atrophies of small hand muscles.
Sex: F, female; M, male; n.d., no data; LL, lower limbs; UL, upper limbs.
Electrophysiological findings in all available mutation carriers are summarized in Table 3. Electrophysiological values are more abnormal in affected men than in women, with usually nonrecordable values especially in the legs compared with the women. Additional symptoms were present only in one case: a patient with p.Leu9Phe mutation was found to have hearing impairment in the left ear, probably not related to CMTX. The pedigrees of reported families are shown in Figure 1—patients marked with numbers were examined in our laboratory. Mutations p.Leu9Phe and p.Leu212Phe were previously reported by us in an abstract form (Seeman et al., 2004).
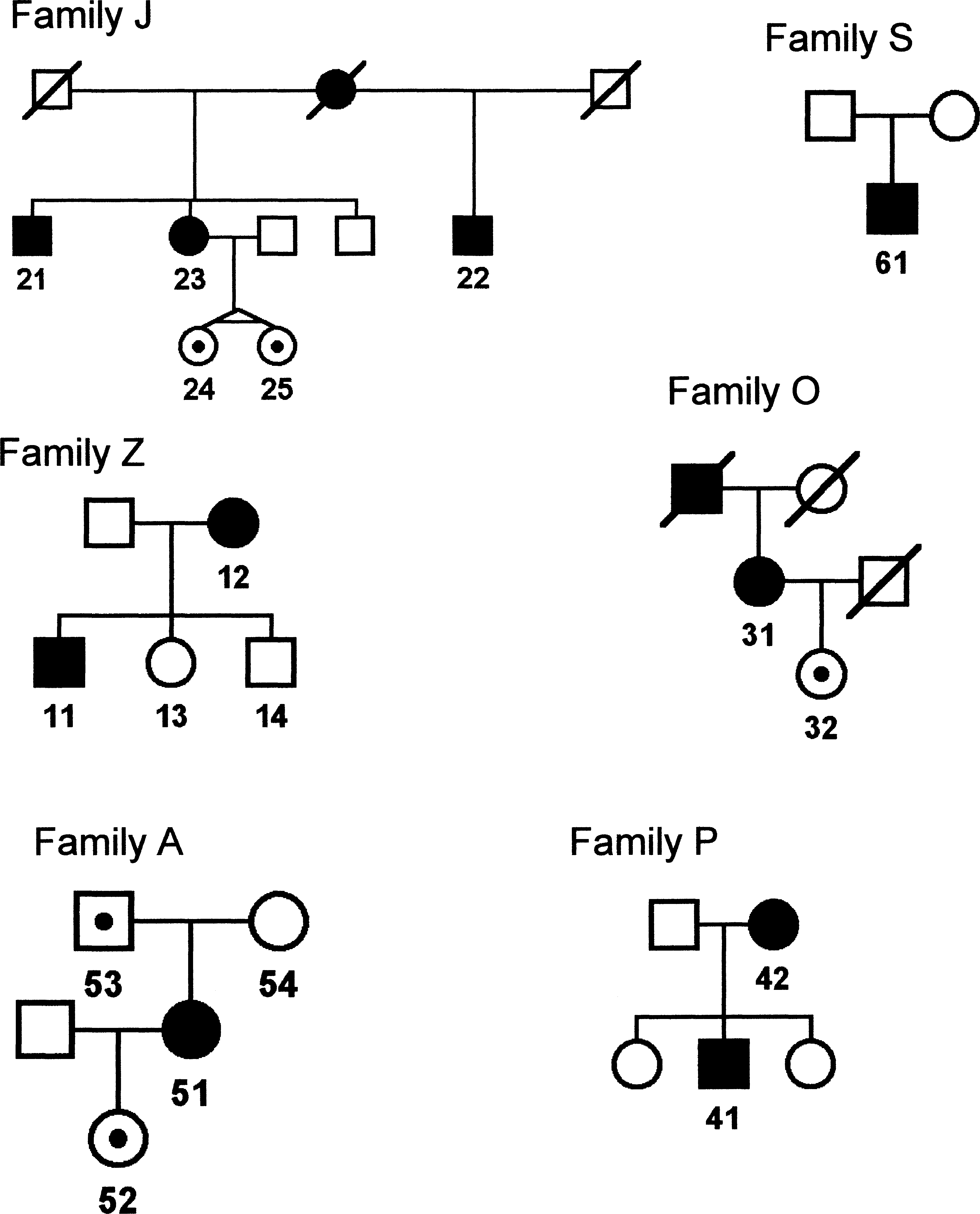
Pedigrees of the reported families. Square, man; circle, woman; black filled icon, affected patient; icon with black dot, healthy member with mutation; slash over the icon, deceased member. The numbers indicate individuals who were examined.
NA, not available; nr, not recordable.
Discussion
Results of this study showed phenotypes associated with six new GJB1 mutations.
The amino acid substitution on the position p.Met1Arg present in family Z is the first mutation affecting the start codon of the GJB1. Deletions of the whole GJB1 gene were already described (Ainsworth et al., 1998; Hahn et al., 1999; Lin et al., 1999; Takashima et al., 2003). We assume that the amino acid change of the first position might result in an alternative initiation of translation at the second in-frame ATG-localized 34 codons downstream. The patient was seen with severely affected lower and upper limbs with bilateral ankle surgery. The clinical and electrophysiological status of his mother shows milder symptoms of CMTX.
The p.Val63Phe amino acid substitution present in the extracellular domain 1 was not previously described. There was another amino acid substitution (Val63Ile) described affecting the same codon of GJB1 (Fairweather et al., 1994; Bone et al., 1997; Matsuyama et al., 2001; Takashima et al., 2003; Casasnovas et al., 2006). Both extracellular domains of the GJB1 gene have the role of connexon-connexon interactions, and therefore the pathology of this mutation can be in the malfunction of these interactions. The patient with p.Val63Phe mutation was seen with mild symptoms of CMT, with age of onset in the second decade. His mother had mild symptoms at the age of 54 years, although subjectively she felt healthy.
Five members of family J with p.Leu9Phe substitution were seen with the mutation. The onset of symptoms started in the first and second decade among the men and in the fourth decade in one woman. The twin sisters from this family were seen with only subclinical symptoms of CMT such as decreased or absent tendon reflexes and discrete electrophysiological abnormalities (Table 3, patient J-25).
The de novo mutations in GJB1 are very rare (Meggouh et al., 1998; Di Iorio et al., 2000). We present the de novo mutation Leu212Phe located in the fourth transmembrane domain of the GJB1 gene. This amino acid substitution was not present in the patient's mother. The maternity was tested by a set of seven microsatellite markers. In addition, the clinical and electrophysiological examination of both parents was normal. The patient has no siblings. This mutation affects a conserved codon of GJB1 and the PANTHER analysis shows pathogen effect with a pdeleterious value of 0.97631. Therefore we believe that all these data support the pathogenic and causal character of this mutation even without the possibility of segregation in the family.
The new reported mutation p.Val170Ile was found in an affected female with mild symptoms of CMT. This mutation is probably a nonpathogenic polymorphism, because the same mutation was detected in her healthy father, who has normal neurological status and normal results of nerve conduction study. We did not find the mutation in her mother. Valin and isoleucin are similar amino acids from the same group. In addition, the Val170 amino acid position is not strongly conserved among species (Fig. 2). The subPSEC value is −3.19634, which is very close to the cutoff value −3 for sequence polymorphism and is much higher than the other five mutations. Val170Ile is the first polymorphism among GJB1 mutations resulting in amino acid change. Almost every mutation so far reported in GJB1 is pathogenic (www.molgen.ua.ac.be/CMTMutations/).

Sequence alignment of gap junction beta 1 proteins from different species. The position of mutation is emphasized with rectangle and the amino acid position with number.
The woman affected with p.Ser17Tyr mutation started to have symptoms in the second decade. The patient's daughter remained asymptomatic because of her low age. The pedigree of family O includes a deceased affected man. His genotype therefore could not be confirmed with DNA analysis. We still assume that this is a causal pathogenic mutation with respect to Panther analysis and conserved Ser17 amino acid position among species (Fig. 2).
Our report should help to further delineate the knowledge about CMTX1 phenotype and the spectrum of GJB1 mutations and to improve genetic counseling in other patients with CMTX1.
Footnotes
Acknowledgments
The authors thank J. Kofer, M.D. (family J), M. Majer, M.D. (family O), Prof. E. Seemanová, M.D. (family S), and I. Štěpánová, M.D. (family J), for sending us the families for examination. The project was supported by a grant VZ 00064203/6506 and GAUK No. 309.
Disclosure Statement
No competing financial interests exist.