
Abstract
Inosine 5′-monophosphate dehydrogenase (IMPDH), which catalyzes a key step in the de novo biosynthesis of guanine nucleotide, is mediated by two highly conserved isoforms, IMPDH1 and IMPDH2. In this study, IMPDH2 genetic polymorphism was investigated in 96 individuals of Caucasian origin. Four single-nucleotide polymorphisms were identified, comprising one previously described single base-pair substitution in the close vicinity of the consensus donor splice site of intron 7 (IVS7+10T>C), and three novel polymorphisms, one silent substitution in exon 9 (c.915C>G), one single base-pair insertion (g.6971_6972insT) within the 3′-untranslated region of the gene, and one substitution located in the promoter region (c.−95T>G) in a transcription factor binding site CRE(A) (cyclic adenosine monophosphate [cAMP] response element). Considering the nature and location of this latter polymorphism, its functional relevance was examined by transfecting HEK293 and Jurkat cell lines with constructs of the related region of IMPDH2/luciferase reporter gene. The c.−95T>G mutation leads to a significant decrease of luciferase activity (HEK293: 55% decrease, p < 0.05; Jurkat: 65% decrease, p < 0.05) compared with the wild-type promoter sequence and, therefore, is likely to determine interindividual differences in IMPDH2 transcriptional regulation. These results might contribute to a better understanding of the variability in clinical outcome and dose adjustments of certain immunosuppressors that are metabolized through the IMPDH pathway or that are IMPDH inhibitors.
Introduction
G
The enzymatic activity of IMPDH is composed of two distinct but nearly identical isoforms encoded by two genes, namely IMPDH1 and IMPDH2. Even if they exhibit disparities in terms of length (more than 18 kb for type I gene versus 5.8 kb for type II) and chromosomic localization (7q31.3-q32 vs. 3p21.2-p24.2, respectively), both genes contain 14 highly conserved exons (Glesne et al., 1993; Glesne and Huberman, 1994). The two isoforms are 84% identical at the amino acid level (Collart and Huberman, 1988; Natsumeda et al., 1990) and display substrate affinities, catalytic activities, and Ki values that are virtually indistinguishable (Carr et al., 1993; Hager et al., 1995). Despite the high level of identity, the two isoforms are not mutually redundant. Homozygous deletion of the IMPDH2 gene results in embryonic lethality at day 9 of gestation in mice (Gu et al., 2000), indicating that the activity of IMPDH1 enzyme is unable to substitute for the activity of IMPDH2 enzyme during early embryonic development. Deletion of the type I gene has a milder effect on phenotype (Gu et al., 2003). In addition, while both isoforms are expressed in most tissues, the regulation of the expression of both IMPDH genes differs significantly (Zimmermann et al., 1995; Gu et al., 1997; Jain et al., 2004). The increased IMPDH activity observed in replicating or neoplastic cells is largely due to an increased expression of the IMPDH type II mRNA, whereas the type I gene remains constitutively expressed during cell proliferation or transformation (Nagai et al., 1991; Nagai et al., 1992). Disparities in 5′-regulatory sequences of type I and II IMPDH genes probably explain these divergences. While expression of the IMPDH1 gene is controlled by three distinct promoter sequences in a tissue-specific manner (Gu et al., 1997), the IMPDH2 gene is regulated by a single promoter (Zimmermann et al., 1995), which contains various transcription factor-binding sites, such as tandem CRE motifs, a Sp1 site, an overlapping Egr-Sp1 site, and a palindromic octamer sequence. These motifs bind with nuclear factors identified as candidate mediators of growth-regulated by IMPDH2 gene expression (Zimmerman et al., 1997).
At the molecular level, IMPDH1 genetic polymorphism has been analyzed in a large extent and numerous missense mutations had been identified as responsible for autosomal dominant retinitis pigmentosa (Kennan et al., 2002; Bowne et al., 2006). Recently, IMPDH2 has generated less interest and only a few IMPDH2 polymorphisms have been described so far, especially a missense substitution, Leu263Phe, likely associated with an altered enzyme activity (Wang et al., 2007), and an intronic polymorphism (IVS7+10T>C) potentially associated with biopsy proven acute rejection in kidney transplantation (Grinyó et al., 2008). The objective of this study was to analyze the full extent of IMPDH2 polymorphism and to explore the in vitro consequences of potentially relevant polymorphisms. We screened all the 14 IMPDH2 exons, their 5′- and 3′-splice site consensus sequences, as well as the described 5′-regulatory region, in genomic DNAs from 96 individuals of Caucasian origin. Four single-nucleotide polymorphisms (SNPs) were identified and the functional impact of one of them, located in the promoter region, was assessed.
Material and Methods
DNA samples
DNA samples from 96 healthy French volunteers of Caucasian origin were collected after approval form the ethical committee and informed consent from each individual was obtained. Total genomic DNA was isolated from peripheral leukocytes using the Nucleon BACC3 kit (Amersham Pharmacia Biotech, Saclay, France), according to the manufacturer's instructions.
Single-strand conformational polymorphism analysis and sequencing
Sixteen different polymerase chain reactions (PCRs) were performed on each DNA sample from the 96 French Caucasians to individually amplify the 14 exons of the IMPDH2 gene, including their 5′- and 3′-splice site consensus sequences, and two sequences spanning the promoter region. Specific primers were designed based on the published nucleotide sequence of the gene (GenBank L39210.1). The name, sequence, and location of primers are listed in Table 1. Each amplification reaction was carried out in a total volume of 25 μL of 10 mM Tris-HCl buffer (pH 8.4) containing 50 mM KCl, 0.2 mM of each dNTP, 0.4 μM of each primer, 200 ng DNA, and 0.6 U Taq DNA polymerase (Applied Biosystems, Foster City, CA). MgCl2 concentration, annealing temperature, and number of cycles were optimized for each primer set (Table 1). After an initial denaturation step at 94°C for 120 s, 30-34 cycles of 60 s at 94°C, 90 s at an optimized annealing temperature, and 90 s at 72°C were carried out. A final extension period of 7 min was performed at 72°C. Size and specificity of PCR fragments were controlled on 1% agarose ethidium bromide-stained gels.
PCR, polymerase chain reaction; F, forward; R, reverse; SSCP, single strand conformational polymorphism.
Nonisotopic single strand conformational polymorphism (SSCP) analysis of PCR fragments was performed using a Genephor Electrophoresis Unit (Amersham Pharmacia Biotech), according to the manufacturer's instructions. PCR products were diluted (1:1) with formamide containing 0.05% bromophenol blue and 0.05% xylene cyanol and subjected to a denaturation step at 95°C for 4 min, followed by rapid cooling on ice. Samples were then loaded into the wells of a precast 12.5% polyacrylamide gel (Gene Gel Excel 12.5/24) for nondenaturing electrophoresis. The conditions used to obtain the clearest and sharpest signals for the different fragments are reported in Table 1. The gels were stained using the DNA Silver Staining Kit (Amersham Pharmacia Biotech). To avoid erroneous shift of electrophoretic mobility due to mutations resulting from possible replication errors of the DNA polymerase, the PCR-SSCP procedure was repeated twice when a sample displayed a SSCP pattern different from that of a reference sample with a wild-type sequence. Nucleotide sequences were determined using an automated DNA sequencer (ABI Prism® 3130 Genetic Analyzer; Applied Biosystems). Fragments were amplified by PCR with primers used for the PCR-SSCP strategy and labeled with the BigDye® Terminator v3.1 kit (Applied Biosystems).
Genotyping of the g.3375C>T polymorphism
To allow rapid genotyping of the g.3375C>T (Leu263Phe) polymorphism described by Wang et al. (2007), a PCR-restriction fragment length polymorphism-based assay was performed. After amplification with IMPDH2-7F and IMPDH2-7R primers (Table 1), the PCR products were digested with the endonuclease Cac8I (New England Biolabs, Ipswich, MA), which cleaves the wild-type allele into two fragments (206 and 63 bp), but leaves uncut the g.3375T allele (269 bp). The digested PCR fragments were then analyzed on a 2% agarose ethidium bromide-stained gel.
Construction of luciferase reporter gene plasmid
A genomic DNA sample from an individual heterozygous for the c.−95T>G substitution was used as a template to amplify the promoter region (−493 to +28) of the IMPDH2 gene. PCR was performed using the sense primer LucF 5′-CTT AAA
Transient transfection assays
Analysis of the effect of the c.−95T>G variant on the promoter activity of a reporter gene was performed by transient transfection assay in the human kidney cell line HEK 293 using the lipofectamine reagent (Invitrogen), according to the manufacturer's recommendations, and in Jurkat lymphocytes by electroporation (Easyject+, 250 V, capacitance: 1500 μm; Eurogentec, Seraing, Belgium). A pSV-β- galactosidase control vector (Promega) was cotransfected with each pGL3 construct to normalize the transfection efficiency.
Cell extract preparation and luciferase activity measurement
Thirty-six hours after transfection, cells were disrupted with the reporter lysis buffer (Promega) and thermic shocks. Luciferase activity was measured using a Berthold Lumat LB 9501 luminometer in 100 μL reagent buffer that contains 25 mM Tris-phosphate (pH 7.8), 8 mM MgCl2, 1 μM dithiotreitol, 0.5% Triton X-100, 18% glycerol, 16 mM ATP, and 2 mM luciferine (Promega). Transfection and reporter assays were repeated in two independent experiments, each experiment comprising three replicates. Statistical analyses were performed using analysis of variance and Turkey's post hoc test. A value of p < 0.05 was considered statistically significant.
Results
Identification of SNPs in the human IMPDH2 gene
To detect sequence variations in the IMPDH2 gene, a PCR-SSCP strategy was developed and applied to genomic DNAs from 96 unrelated individuals of Caucasian origin. One DNA sample, obtained from a homozygote for a wild-type allele of IMPDH2, as confirmed previously by sequencing, was used as a reference sample for SSCP analysis. Of a total of 96 DNA samples, 65 were identified as homozygous for an apparent wild-type IMPDH2 allele (Fig. 1). In contrast, 31 samples showed distinctive mobility shifts in at least one of the 16 PCR products, indicating the presence of polymorphism(s) in the corresponding region (Fig. 1). Sequencing of these fragments allowed the characterization of four different polymorphisms. Their frequencies are reported in Table 2.
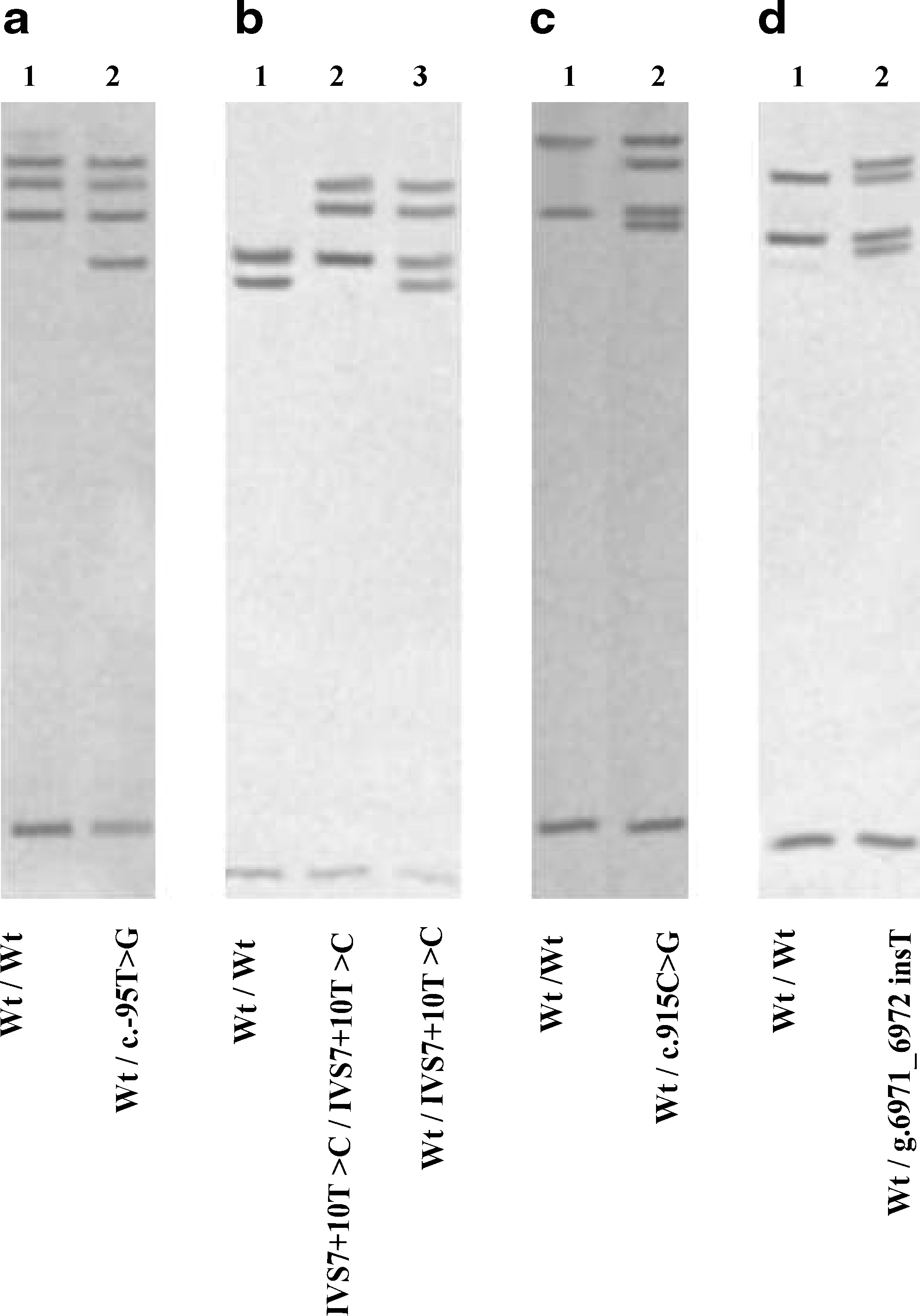
PCR-SSCP analysis of the human IMPDH2 gene. The 14 exons with their splice site junctions and the promoter region of IMPDH2 were separately amplified from genomic DNAs from 96 individuals of Caucasian origin. The PCR products were then subjected to electrophoresis in nondenaturing gels. One DNA sample with a wild-type genotype (lanes 1, wt) was used as a reference sample for SSCP analysis. Abnormal SSCP patterns were observed for the promoter region (
Number of alleles.
The first abnormal profile (Fig. 1a) corresponds to a nucleotide substitution within the 5′-flanking region of the gene at position c.−95 (transition T−95 to G). The second polymorphism (Fig. 1b) corresponds to a previously described SNP located in close vicinity of the consensus donor splice site of intron 7 (IVS7+10T>C, rs11706052). This SNP was found in a homozygous state in one individual (Fig. 1b, lane 2). The third polymorphism was detected in exon 9 (Fig. 1c) and corresponds to a silent substitution (c.915C>G, p.Val305Val). The last polymorphism was identified downstream of exon 14, in the 3′-UTR of the gene (Fig. 1d). It consists of a T insertion located 1 bp upstream of the polyadenylation site AATAAA (g.6971_6972insT).
The analysis of the distribution of polymorphisms described earlier allowed the characterization of five different IMPDH2* alleles in our population. They comprise the wild-type allele, IMPDH2*1, which is the most frequent (83.4%), and four alleles that we termed IMPDH2*1B, IMPDH2*2, IMPDH2*3, and IMPDH2*4. These novel alleles have been submitted to GenBank with accession numbers AY491523, AY491521, AY491522, and AF491524, respectively. The IMPDH2*1B, IMPDH2*2, IMPDH2*3, and IMPDH2*4 alleles carry the polymorphisms c.915C>G, c.−95T>G, IVS7+10T>C, and g.6971_6972insT, respectively.
As the g.3375C>T substitution, corresponding to the Leu263Phe polymorphism described by Wang et al. (2007), was not detected in our Caucasian population by PCR-SSCP analysis, a PCR-restriction fragment length polymorphism assay, using the Cac8I restriction enzyme, was performed and applied to all the PCR products first analyzed by PCR-SSCP (data not shown). None of the tested samples harbored this substitution, which confirmed that our PCR-SSCP strategy did not fail to identify this substitution and that the frequency of this mutation is quite low.
Functional significance of the c.−95T>G promoter polymorphism
The promoter activities of IMPDH2 promoter/luciferase reporter gene constructs, harboring either the wild-type or the c.−95T>G containing sequence, were assessed by transient transfection assays in both HEK293 and Jurkat cell lines. The promoterless pGL3-Basic vector was used as the baseline control (data not shown) and the pGL3-Wt luciferase activity was used as a 100% reference. In both cell lines and in all experiments, the plasmid containing the wild-type fragment yielded luciferase activity which was about 100-fold higher than that of the empty vector. As seen in Figure 2A, in HEK293 cells, a significantly lower luciferase activity (55% decrease; p < 0.05) was observed for the construct containing the c.−95T>G polymorphism, when compared with the wild-type construct. In Jurkat lymphocytes (Fig. 2B), the same trend was observed for the c.−95T>G variant with a significant decrease in the promoter activity of 65% (p < 0.05), compared with that of the wild-type construct.

Promoter activity of the wild-type and c.−95T>C variants in HEK293 (
Discussion
In this study, we developed a simple and efficient strategy based on a PCR-SSCP procedure to screen for sequence variations of the IMPDH2 gene in a Caucasian population. This method had been used for the detection of SNP in several genes (Marez et al., 1997; Lo-Guidice et al., 2002; Cauffiez et al., 2004). Because the sensitivity of SSCP is closely related to the electrophoresis conditions, the analysis was performed after optimization of multiple parameters affecting the single-strand separation, such as time, temperature of electrophoretic migration, and gel matrix (Sheffield et al., 1993). Under optimal experimental conditions, we observed clear differences in band patterns when a PCR product was different from the wild-type sequence. However, despite many precautions, some polymorphisms might have not been detected under the chosen conditions.
Our study has allowed the characterization of three novel polymorphisms that correspond to one silent SNP in exon 9 (c.915C>G), one nucleotide substitution in the promoter region (c.−95T>G), and one insertion (g.6971_6972insT) in the 3′-UTR of the IMPDH2 gene. In addition, the previously described intronic substitution, IVS7+10T>C, was also identified (Wang et al., 2007). These polymorphisms define at least four allelic variants of IMPDH2, namely IMPDH2*1B, IMPDH2*2, IMPDH2*3, and IMPDH2*4. However, additional alleles resulting from combination of these polymorphisms might exist, as one individual was identified as heterozygous for both IVS7+10T>C and c.915C>G substitutions. The three alleles IMPDH2*1B, IMPDH2*2, and IMPDH2*4, which carry the c.915C>G, c.−95T>G, and g.6971_6972insT, respectively, are rare variants with a frequency ≤1.0% in our population, whereas the fourth allelic variant, IMPDH2*3 (IVS7+10T>C), is more frequent, with an allelic frequency of 14.6% in the same population.
Considering their nature and location, the c.915C>G and g.6971_6972insT polymorphisms are unlikely to have any functional consequences. The clinical significance of the IVS7+10T>C polymorphism remains unclear, because no functional analysis had been previously performed to our knowledge. Nevertheless, this polymorphism has been recently associated with approximately three-times odds of biopsy proven acute rejection in a cohort of 237 renal transplant recipients (Grinyó et al., 2008).
More interestingly, the c.−95T>G substitution is located in a CRE(A) motif (
It is interesting to speculate on the possible functional consequence of the c.−95T>G polymorphism in vivo. If interindividual variations in the capacity to express IMPDH2 exist, they could then be reflected by differences in therapeutic response to immunosuppressive agents that interrupt IMPDH2 pathway. As an example, mycophenolate mofetil (MMF, CellCept®, Roche, Neuilly sur Seine, France) is the prodrug of mycophenolic acid, an inhibitor of IMPDH that has been approved for the prevention of acute rejection in kidney, heart, and liver transplantations (Allison and Eugui, 1993). Mycophenolic acid is a fivefold more potent inhibitor of the type II isoform of IMPDH than of the type I isoform (Carr et al., 1993; Allison and Eugui, 2000). Further, a substantial heterogeneity in drug response has been observed among patients treated with MMF (Budde et al., 2000; Glander et al., 2001). Because IMPDH2 gene expression is in part transcriptionally regulated, individuals who harbor the c.−95T>G polymorphism might have diminished IMPDH2 gene transcription, and therefore abnormal clinical response to treatment targeting IMPDH2 pathway, such as MMF. Genotyping of patients under MMF treatment could be of interest to investigate the possible implication of this promoter polymorphism in MMF therapy efficiency.
As it is recognized that IMPDH2 is a critical enzyme in the metabolic pathway of the thiopurine drugs, that is, azathioprine and its metabolite 6-mercaptopurine, because of its role in the synthesis of the active metabolites or 6-thioguanines, the analysis of IMPDH2 as a candidate gene for abnormal response to thiopurines remains to be further evaluated. Indeed, even if genetically determined level of the thiopurine S-methyltransferase enzyme is known to modulate the therapeutic efficacy and toxicity of the thiopurine drugs (McLeod and Siva, 2002), thiopurine S-methyltransferase deficiency only explained around 30% of cases of myelosuppression following 6-mercaptopurine or azathioprine therapy (Black et al., 1998; Ishioka et al., 1999; Naughton et al., 1999; Colombel et al., 2000; Dubinsky et al., 2000). The possibility arises that thiopurine drug toxicity consequent to altered IMPDH2 gene expression could be important, and so it warrants further investigation.
In conclusion, our data provide evidence that the IMPDH2 gene is polymorphic. Despite its low frequency, the c.−95T>G promoter polymorphism is likely to disrupt the transcription factor binding site CRE(A) and may result in interindividual differences in IMPDH2 transcriptional regulation and, consequently, in interindividual differences in IMPDH2 expression. Further, these data could be helpful to study the relationship between a given patient's clinical response toward a treatment that modifies the IMPDH2 pathway and its genotype at the IMPDH2 locus.
Footnotes
Acknowledgments
This study was supported by the Centre Hospitalier Régional et Universitaire de Lille, the Université Lille Nord de France, and the Conseil Regional Nord-Pas de Calais. The authors thank Anne Engels for her excellent technical assistance.
Disclosure Statement
No competing financial interests exist.