
Abstract
Karyotyping was done in 100 children suspected of having chromosomal abnormalities of genetically uncertain syndromes, multiple congenital anomalies, short stature, dysmorphic features, unclassified mental retardation, and Down syndrome. A total of 56 patients had an abnormal karyotype: ring chromosome of 13 was seen in 1 patient (1.78%), and trisomy 21 was seen in 29 patients (51.78%) who were diagnosed as Down syndrome patients. Among them, 9 were male patients (31.03%) (47,XY+21) and 18 were female patients (47,XX+21) (62.06%); 2 patients showed 47,XY+21/46,XY (mosaicism) (6.89%). Chromosomal rearrangements involving chromosome numbers 13, 14, and 21 were seen in three patients. Among them, one patient had t(13;21) [45,XX,t(13;21)] and two patients had 45,XY,t(14;21). Trisomy 22 was seen in three patients (5.3%), marker chromosome was seen in two patients (3.57%), 46,XY,16qh variant was seen in one patient (1.78%), 46,XX,der(2) was seen in one patient (1.78%), 46,XX,14ps+ was seen in two patients (3.57%), and 46,XY,r(18) was seen in three patients (5.37%). Apart from this, 11 patients (19.64%) had sex chromosome aberrations: 45,XO was seen in 3 patients (27.7%), 4 patients were mosaic for Turner syndrome (45,XO/46,XX) (36.36%), and 4 patients had 46,Xi(Xp) (36.36%), and the remaining 44 patients had normal karyotypes. All of them showed phenotypic-cytogenetic heterogeneity. These findings suggest that cytogenetic analysis is useful in the investigation of children with genetic disorders of unknown origin to confirm clinical diagnosis and to allow for proper genetic counseling.
Introduction
C
The impact of chromosomal abnormalities is greatest during fetal life when they have their highest frequency and represent a major cause of fetal loss (Seashore and Wappner, 1996). The frequency of various chromosomal abnormalities is quite different in neonates (0.7%) when compared with abortuses (about 50%), because some aneuploidies are lethal in utero (Thompson et al., 1991).
The major autosomal abnormalities share a number of phenotypic features that are not distinctive or specific, including mental retardation (MR), cardiac malformation, and growth deficiency. Although there is variability within every cytogenetic syndrome, neonatal death and serious congenital malformations are frequent manifestations. Most of the specific cytogenetic syndromes have a constellation of features that distinguish them and allow the clinician to suspect the condition (Mokhtar, 1997).
Several studies have shown documented chromosomal abnormalities among unselected populations of neonates and older children (Hook and Hamerton, 1977). Other cytogenetic studies among selected populations with abnormal phenotype features have also been conducted (Singh, 1977; Verma and Dosik, 1980). The frequency of chromosomal abnormalities is known to be significantly higher in selected populations than in unselected populations (Shah et al., 1990; Nkanza and Tobani, 1991).
Among the subjects examined, we found a number of patients with varying genetic problems, and then our special effort has been extended to deal with genetic counseling for them. The situation emphasized the need for cytogenetical studies in them to enquire into these problems and also to contribute on chromosomal basis to clinical diagnosis of congenital malformations. One-hundred cases with congenital defects were subjected to chromosome analysis. This report presents a summary of clinical examinations, together with the chromosomal findings in those 100 cases.
The aims of this study were to investigate the different types of chromosomal aberrations and their relative frequencies in a group of children with suspected genetic disorders and to identify precisely the role of cytogenetic investigation in confirming the diagnosis, thus allowing proper genetic counseling to be offered.
Materials and Methods
The study included 100 children with various phenotypic abnormalities such as genetically uncertain syndromes, multiple congenital anomalies, short stature, dysmorphic features, unclassified MR, Down syndrome, and Turners syndrome. Their ages ranged from 1 month to 15 years. They were referred by the outpatient block of the Pediatrics Department, Indraprastha Apollo Hospitals, New Delhi, and Apollo Health City, Hyderabad. All the patients were subjected to complete physical examination, and family history along with pedigree was taken to exclude known nonchromosomal causes of anomaly. Cytogenetic analysis was carried out for all the patients in the Molecular Biology and Immunology Lab at Indraprastha Apollo Hospitals, New Delhi, and the Molecular Biology and Cytogenetics Lab at Apollo Hospitals, Hyderabad. The study included peripheral lymphocyte culture by a standard method using the G-banding technique on the level of 400 bands according to Seabright (1971). At least 30 metaphases were scored for each patient. Three cells were karyotyped according to International System for Human Cytogenetic Nomenclature (ISCN) criteria (Mitelman, 2005). Usually the total chromosome count was determined in 30 cells, but if mosaicism was suspected then 50 or more cell counts were undertaken (Kingston, 1994).
Results
Of the 100 patients on whom chromosomal analysis was done, chromosomal aberrations detected in 56 patients involved autosomes, whereas only 11 patients involved sex chromosomes (Table 1). A total of 56 had an abnormal karyotype: ring chromosome of chromosome number 13 was seen in 1 male patient [46,XY,r(13)] (1.78%) (Fig. 1) who showed partial neck holding, premature birth, low birth weight, and microcephaly, and trisomy 21 was seen in 29 patients (51.78%) who were diagnosed as Down syndrome patients (Fig. 2) (Table 2). Among them, 9 were male patients (31.03%) (47,XY+21) (Fig. 2) and 18 were female patients (47,XX+21) (62.06%); 2 patients showed 47,XY+21/46,XY (mosaicism) (6.89%). These children with trisomy 21 showed brachycephaly, epicanthic folds, flat nasal bridge, mongloid facial features, folded or dysplastic ears, small ears, open mouth, protruding tongue, abnormal teeth, short neck, excessive skin at nape of the neck, and space between the first and second toes (sandal gap deformity). Chromosomal rearrangements involving chromosome numbers 13, 14, and 21 were seen in three patients. Among them, one patient had t(13;21) [45,XX, t(13;21)] and two patients had 45,XY, t(14;21) (Figs. 3 and 4) (Table 1). These children with Robertsonian translocation t(13;21), t(14;21) showed partial neck holding, premature birth, low birth weight, microcephaly, flat facial profile, hypotonia, sandle sign, brachydactyly, and hole in the heart. Trisomy 22 was seen in three patients (5.3%), and marker chromosome (47,XX+mar) was seen in two female patients (3.57%) (Fig. 5). Children with marker chromosome showed delayed speech, delayed milestones, partial neck holding, muscular weakness, premature birth, and general hypotonia. 46,XY,16qh variant was seen in one male patient (1.78%), and 46,XX,der2 was seen in one female patient (1.78%) who had delayed speech, delayed milestones, muscular weakness, premature birth, general hypotonia, and hyperplaxia. 46,XY,14ps+ was seen in two male patients (3.57%) (Fig. 6) who were obese and showed delayed milestones. A ring chromosome of chromosome number 18 [46,XY,r(18)] was seen in three male patients (5.37%) (Fig. 7) who showed partial neck holding, premature birth, low birth weight, microcephaly, and delayed milestones.

A partial male karyotype showing a ring chromosome of chromosome 13.

A partial male karyotype showing trisomy 21.

A partial female karyotype showing Robertsonian translocation t(13;21).

A partial female karyotype showing Robertsonian translocation t(14;21).
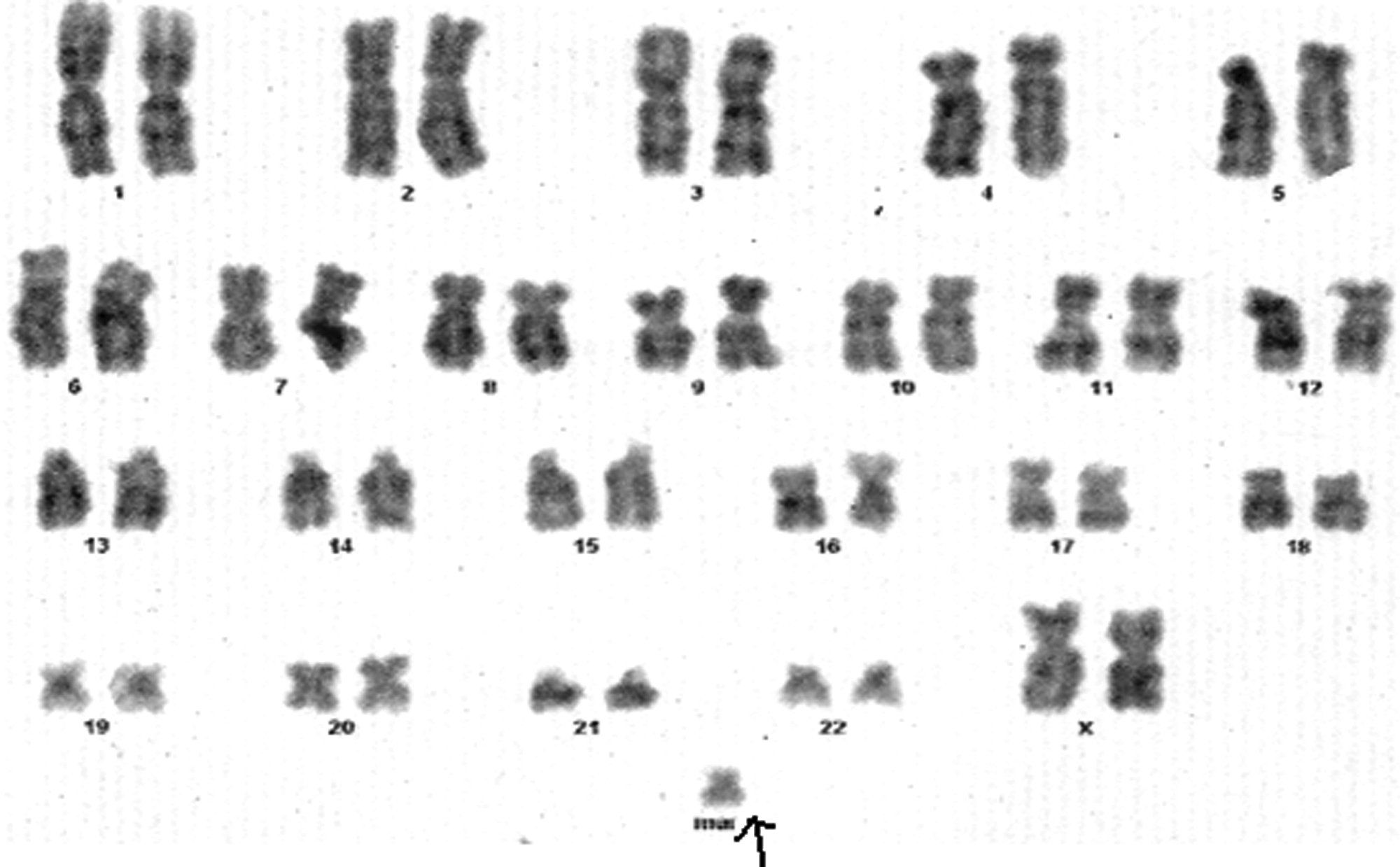
A female karyotype showing a marker chromosome (arrow).
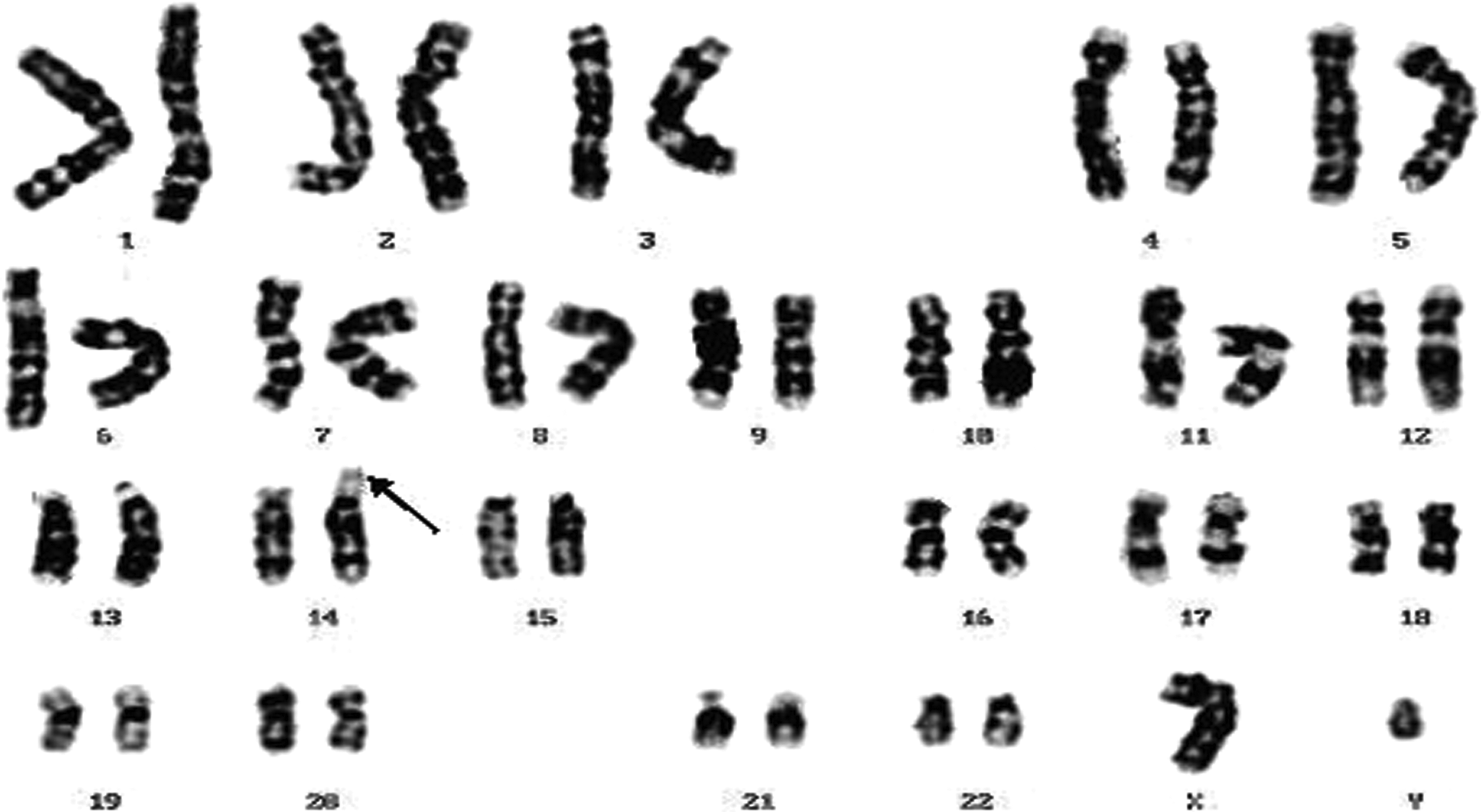
A male karyotype showing 14ps+ (arrow).

A male karyotype showing a ring chromosome of chromosome 18 (arrow).
Apart from this, 11 patients (19.64%) had sex chromosome aberrations; 45,XO was seen in 3 patients (27.7%) (Fig. 8), 4 patients were mosaic for Turner syndrome (45,XO/46,XX) (36.36%) (Table 1), and 4 patients had 46,Xi(Xp) (36.36%) (Fig. 9) and the remaining 44 patients had normal karyotypes. Children showing a loss of X (46,XO) chromosome (Turners syndrome) showed premature birth, obesity, and webbed neck.
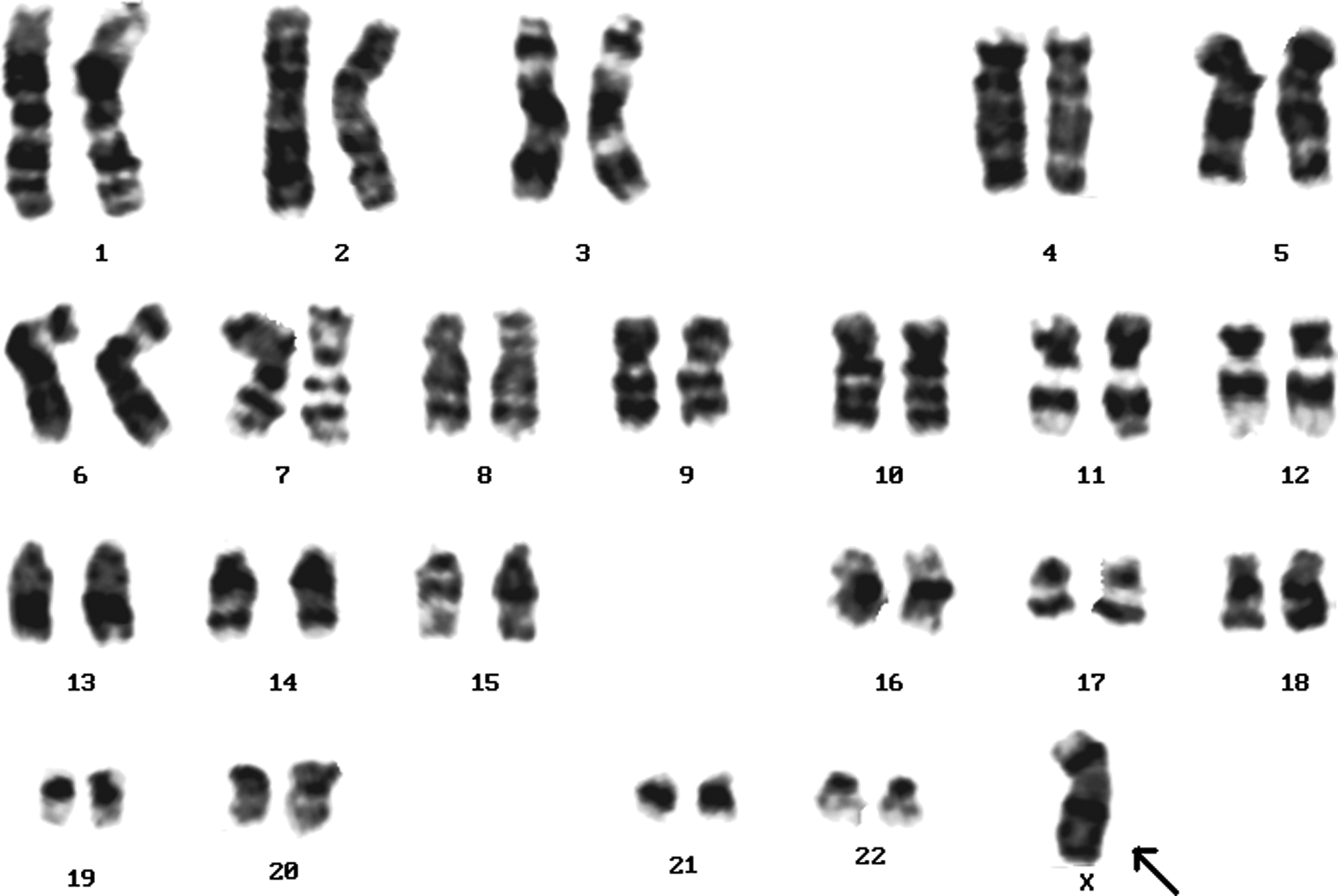
A female karyotype showing a loss of X chromosome (Turners syndrome) (arrow).

A female karyotype showing isochromosome of X chromosome i(Xp) (arrow).
Discussion
In consecutive neonatal studies, autosomal anomalies are usually as common as sex chromosome anomalies (Gardner and Sutherland, 1996). In studies based on a referred population with phenotypic abnormalities, such as this study, autosomal anomalies (78.5%) (Table 2) are much higher than those of the sex chromosomal anomalies (19.64%). This figure is in agreement with other surveys (Al-Awadi et al., 1985; Berry et al., 1991). This is mainly due to the fact that sex chromosome imbalance has a much less deleterious effect on the phenotype than does autosomal aneuploidy (Gardner and Sutherland, 1996).
Different investigators have reported wide variations in the frequency of chromosomal aberrations in individuals suspected of having genetic disorders (Verma and Dosik 1980; Shah et al., 1990). Berry et al. (1991) studied 114 patients and found chromosomal aberrations in 18 patients (15.8%). Navsaria et al. (1993) along with other investigators evaluated patients and found chromosomal aberrations in 12-16% of the patients (Al-Arrayed, 1991; Al-Awadi et al., 1992).
The new technical approaches have proved essential for understanding of chromosome aberrations relevant to congenital disorders, thus promoting the development of medical cytogenetics. Evidence has been presented to show that genetic counseling should be made in close association with cytogenetic examination. In India, however, a system combining genetic counseling directly with cytogenetic investigation has not been well established at present. In the present cytogenetic survey, 100 cases diagnosed as congenital defects were chromosomally investigated, and about 56% of the cases displayed chromosomal defects. A similar higher frequency (40%) of chromosomal abnormalities was reported by Kenue et al. (1995) among 120 patients.
Trisomy 21 has been recognized for more than 100 years. Because it is a common and familiar disorder, Down syndrome has been studied much more thoroughly than other chromosomal disorders. The Down syndrome phenotype is due to trisomy 21 (Gardner and Sutherland, 1996). The frequency of Down syndrome in patients with abnormal chromosomes in this study was 57% (Table 1). This value was similar to other surveys (Kenue et al., 1995; Gardner and Sutherland, 1996). This could be attributed to its easy detection at the clinical level.
The frequency of standard trisomy 21 amongst Down syndrome patients in this study was 84.37% (27 patients). This value is in agreement with other surveys, which ranged from 84.6% to 95% (Gardner and Sutherland, 1996). The frequency of mosaicism in Down syndrome patients is reported to vary between 0% and 4%. Only 6.25% of patients (two patients) with Down syndrome in this study had mosaic Down syndrome. The frequency of translocation involving chromosome 21 among three patients in this study was 9.37%. This figure is higher than the previously reported values [5.6% (Wright et al., 1990; Pullian and Huether, 1986), 5.2% (Mikkelsen et al., 1990), 6.81% (Mutton et al., 1996)], but the actual level depends on the maternal age distribution and the rate of indication for prenatal diagnosis (Krishna et al., 1992) (Fig. 2).
Although individually rare, partial autosomal aneuploidies are the second most common chromosomal abnormality after trisomy 21 (Seashore and Wappner, 1996). Our results (3.6%) confirm this finding, but the frequency found in our study is higher than that reported by Kenue et al. (1995) (0.8%) and Al-Awadi et al. (1992) (0.3%). It is believed that excess or loss of several contiguous genes along the chromosome involved will explain the phenotypes of these conditions.
Marker chromosomes are defined as abnormal chromosomes that cannot be fully characterized based on standard cytogenetic analysis (Krishna et al., 1992). The incidence of marker chromosomes has been found to be 0.024% among neonates (Sachs et al., 1987). The association of an additional marker chromosome and abnormal phenotype has been described by Ballesta et al. (1991) in 14 probands with MR and malformations (Webb, 1994). The two patients with a marker chromosome detected in this study had been referred with congenital malformations, but the precise origin of the markers could not be determined using available techniques (Fig. 1). About 37.5% of metaphases screened have shown the marker chromosome in both the cases and the remaining 62.5% have shown normal karyotypes.
The incidence of Turner syndrome in consecutive neonates has been reported to be 0.04% (Navsaria et al., 1993). Turner syndrome is one of the few chromosomal aberrations that can be recognized clinically during infancy or childhood based on short stature, broad shield chest, lymphoedema of the lower limbs, webbed neck, and multiple minor anomalies (Ballesta et al., 1991). However, karyotyping is necessary to confirm the diagnosis. This study included 11 patients (19.64%) who had sex chromosome aberrations, 45,XO was seen in 3 patients (27.7%), 4 patients were mosaic for Turner syndrome (45,XO/46,XX) (36.36%) (Table 1; Fig. 5), and 4 patients had 46,Xi(Xp) (36.36%). This frequency agrees with that reported by Guera et al. (1991) (18%).
Many of the patients (44 patients) had normal karyotypes; however, some of these patients might have submicroscopic rearrangements, which could have been missed by conventional cytogenetic analysis because of its low resolution to pick up the subtle anomalies. Hence, this study further reinforces the necessity of fluorescence in situ hybridization (FISH) investigations as an adjunct to conventional cytogenetic analysis in the characterization of chromosomal abnormalities. As this technique becomes more widely used, more accurate assessment of complex rearrangements can be anticipated (Guera et al., 1991).
It is always a challenge to transfer new technology from a research setting to clinical applications. Three large studies (Vorsanova et al., 1998; Knight et al., 1999; Ballif et al., 2000) have shown that in 3-7% of patients, subtelomeric aberrations can be the cause of unexplained moderate-to-severe MR or developmental delay with dysmorphism. Indeed, if the phenotype is classical for a chromosome aberration and the results with subtelomeric probes are normal, one should consider extending the study to markers covering the entire genome within useful distances (Spikes et al., 1995). Therefore, we consider FISH with multiple subtelomeric probes to be a valuable diagnostic tool that should be implemented in all clinical cytogenetics laboratories. We will primarily plan to detect submicroscopic aberrations in patients with unexplained MR by FISH using multiple subtelomeric probes in the follow-up of this study.
Conclusion
Among a group of children with phenotypic abnormalities, the frequency of autosomal chromosomal aberrations was found to be much higher than sex chromosome anomalies. Trisomy 21 and partial autosomal aneuploidy were the most frequent. The precise delineation of a major autosomal trisomy is only possible using clinical examination and cytogenetic tools. It is further evident from this study that conventional cytogenetics should be included in the routine diagnostic tests for studying the congenital defects in children and should be further supplemented with FISH to study the subtle chromosomal rearrangements, which can be missed by cytogenetic analysis.
Footnotes
Acknowledgments
The authors would like to acknowledge the pediatricians and the patients for their cooperation. We would also like to acknowledge the management of Apollo Hospitals for support in every possible way.
Disclosure Statement
No competing financial interests exist.