
Abstract
Mutations in the GJB2 gene account for up to 50% of hereditary nonsyndromic hearing loss in several populations. Over 200 mutations are already described in this gene, and three of them, c.35delG, c.167delT, and c.235delC, are the most frequent in Caucasians, Ashkenazi Jews, and Asians, respectively. Most of GJB2 hearing loss-related mutations are recessive, but a few dominant alleles have also been described. Apart from the clearly pathogenic mutations, there are some other variants whose pathogenicity is still controversial, such as p.Met34Thr, p.Val37Ile, p.Arg127His, and p.Val153Ile. The p.Arg127His allele has been found in some mono- and biallelic hearing-impaired patients from several countries. In this article we report on some Portuguese patients harboring this mutation. Taking into consideration the analysis of these Portuguese cases as well as the genetic and functional data regarding p.Arg127His available in the literature, we conclude that this variant may be a cause of hearing loss depending on environmental factors and/or genetic background.
Introduction
G
Here we report on three Portuguese families affected with hearing loss, in which we have identified p.Arg127His. This variant is common in both deaf and hearing Indians, presenting allele frequencies of 0.123 and 0.175, respectively, in the study by RamShankar et al. (2003). Similar frequencies were reported by Ramchander et al. (2004) in a different sample of Indian patients and hearing controls. The p.Arg127His variant was also identified in 19.4% of the chromosomes of 54 hearing-impaired Slovak Romany (Gypsies), a people with Indian ancestry (Minárik et al., 2003; Álvarez et al., 2005). Interestingly, two of the families here presented belong to the Gypsy community. We discuss the pathogenicity of p.Arg127His taking into consideration our results as well as the information available in the literature on genetic and functional aspects of this mutation. In this way, this work may contribute to a better understanding of the role of p.Arg127His mutation in hearing loss.
Materials and Methods
Patients
This study focuses on three Portuguese families (coded as BS, DM, and EL), presenting with bilateral nonsyndromic, sensorineural hearing loss, whose analysis was performed when screening Portuguese affected families for GJB2 mutations. All patients were audiologically evaluated by pure tone audiometry. Informed consent was obtained from all the participants.
Genetic analysis
Blood samples were taken from the participants, and genomic DNA was extracted. The GJB2 gene coding region and acceptor splice site was amplified, and sequenced using the newly designed forward primer 2aF 5′-AAGTCTCCCTGTTCTGTCCT-3′ and reverse primer 2bR 5′-GGCATCTGGAGTTTCACC-3′. All the patients, except I:2 from family DM, were further studied in regard to the del(GJB6-D13S1830) and del(GJB6-D13S1854) GJB6 deletions using the same methodology as del Castillo et al. (2005). Afterward, these individuals were also screened by sequencing for mutations in the GJB2 basal promoter, exon 1, and donor splice site, according to Matos et al. (2007).
Results
The hearing-impaired individuals belonging to three Portuguese families were analyzed in respect to GJB2 coding region and acceptor splice site. With the exception of individual I:2 belonging to family DM, these patients were also tested for both the common GJB6 deletions and screened for mutations in the GJB2 basal promoter, exon 1, and donor splice site. The variant p.Arg127His (R127H) was found in the three families analyzed, but in two of them a second GJB2 mutation, p.Trp24X (W24X) or c.-23 + 1G>A (often referred as IVS1 +1G>A), was also identified (Fig. 1). No further GJB2 mutation, or GJB6 deletion, was found.
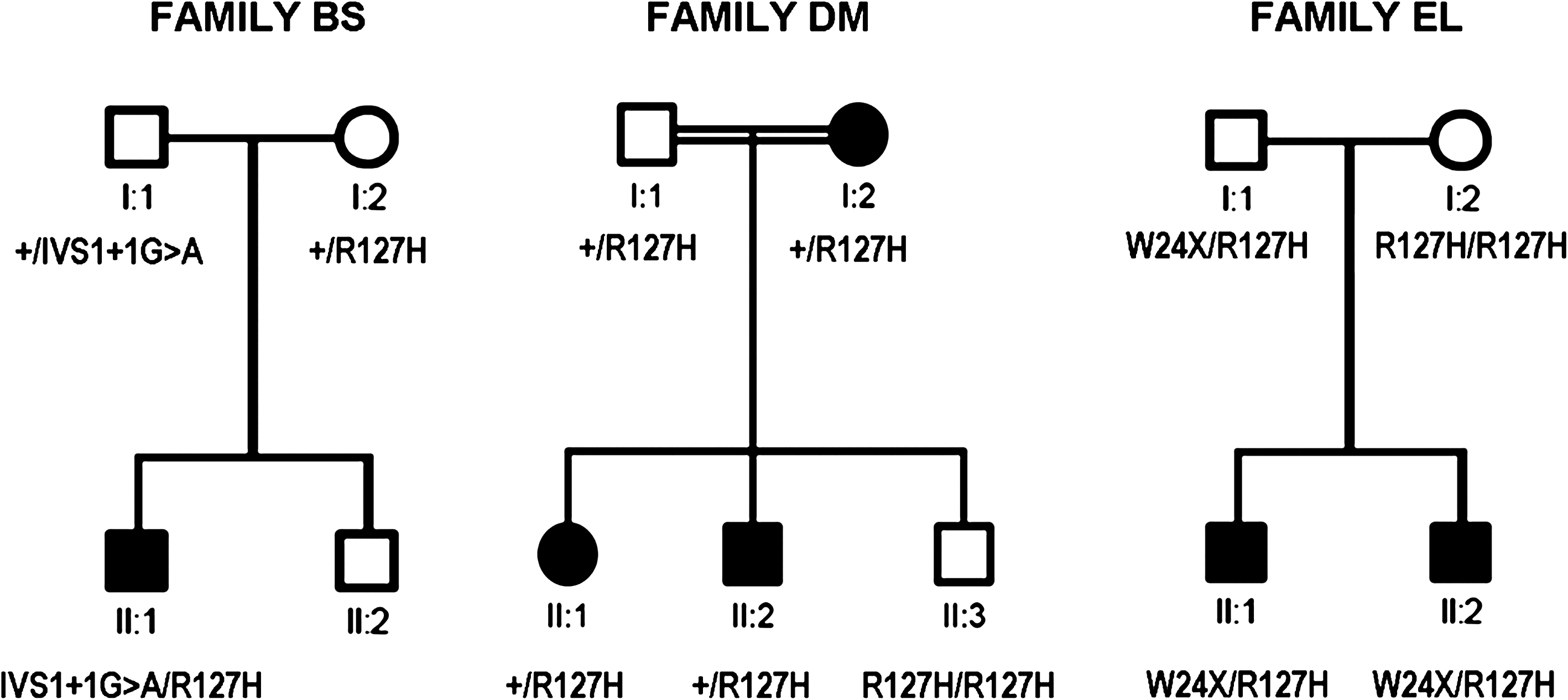
Pedigrees of the three Portuguese families under study, family BS, family DM, and family EL. Black symbols represent hearing-impaired individuals, and open symbols represent normal hearing individuals. W24X and R127H are abbreviations for the p.Trp24X and p.Arg127His mutations, respectively.
In one of the families (EL), of Gypsy ethnicity, the proband, his affected sibling, and the normal-hearing father are compound heterozygous for p.Trp24X/p.Arg127His; the normal-hearing mother is homozygous for p.Arg127His. In the second family (DM), consanguineous and also of Gypsy ethnicity, the p.Arg127His mutation does not seem to segregate with hearing loss since the two affected siblings are heterozygous for p.Arg127His, with no accompanying GJB2 mutation or GJB6 deletion, while their unaffected sibling is homozygous for this mutation. In the third family (BS), of unknown ethnicity, the proband is heterozygous for p.Arg127His as well as for the recessive, donor splice-site mutation c.-23 + 1G>A. Genetic analysis on the parents' DNA was performed to determine whether the two mutations are in trans. It was observed that each parent is a carrier of one of the mutations, so the child is effectively compound heterozygous.
Discussion
We have analyzed three Portuguese families (BS, DM, and EL) presenting with sensorineural hearing loss, and having in common the controversial p.Arg127His mutation in GJB2. However, this variant does not seem to be a cause of hearing impairment in all the three families.
In family DM, the results obtained suggest that the hearing loss is not likely to be related to the p.Arg127His mutation since both affected siblings are heterozygous for this variant, with no accompanying GJB2 mutation or GJB6 deletion, while the normal-hearing sibling is homozygous for p.Arg127His. The cause of the hearing loss might probably lie in recessive alleles of other genes brought together by consanguinity.
In the other two families, BS and EL, the p.Arg127His mutation could be involved in the hearing loss observed, since it occurs in compound heterozygosity with two well-established recessive mutations, p.Trp24X and c.-23 + 1G>A, in the affected individuals (although other gene or environmental factors cannot be excluded as causes for the hearing loss). In family BS, the p.Arg127His variant seems to behave as a recessive allele since the proband is a c.-23 + 1G>A/p.Arg127His compound heterozygote, and the parents, each one a carrier of one of the mutations, have normal hearing. In family EL, the proband and his affected sibling are both compound heterozygous for p.Trp24X and p.Arg127His, a similar situation to the one observed in a hearing loss case reported by Roux et al. (2004). The mutation p.Arg127His has also been found, in other cases of hearing loss, associated with the pathogenic recessive mutation p.Glu47Lys (Prasad et al., 2000), and also with the controversial variants p.Met34Thr (Roux et al., 2004; Snoeckx et al., 2005), p.Val37Ile (Yaeger et al., 2006) and p.Val153Ile (Najmabadi et al., 2005, Santos et al., 2005). Noteworthy, in the study by Roux et al. (2004), the three p.Arg127His alleles identified in the patient cohort occurred in compound heterozygosity with p.Met34Thr (two probands) and with p.Trp24X (one proband), while in the general population no compound heterozygosity involving p.Arg127His was reported. The authors considered significant the fact that compound heterozygosity for p.Met34Thr and p.Arg127His—identified twice among the patients—had not been observed in the general population controls. In other study (Tóth et al., 2004), p.Arg127His was shown to segregate in homozygosity with hearing loss. All these findings would support the pathogenicity of the p.Arg127His variant.
However, some observations reported in the literature seem to argue against the pathogenic nature of p.Arg127His. In family EL and in the case described by Roux et al. (2004) mentioned above, compound heterozygosity for p.Trp24X and p.Arg127His was also observed in normal-hearing relatives. A puzzling behavior of the p.Arg127His variant is again evident when considering the reports of normal-hearing individuals (Marlin et al., 2001) and hearing-impaired individuals (Uyguner et al., 2003) who are compound heterozygotes for the c.35delG and p.Arg127His mutations. Moreover, normal-hearing individuals homozygous for p.Arg127His have been reported (RamShankar et al., 2003; Roux et al., 2004; Dahl et al., 2006), and studies have been published in which the frequency of the p.Arg127His variant in hearing-impaired and normal-hearing individuals is not significantly different, in comparison to clearly pathogenic mutations such as p.Trp24X and c.35delG (RamShankar et al., 2003; Ramchander et al., 2004; Roux et al., 2004).
Given the discrepancies observed in the genetic studies, investigating the functionality of the p.Arg127His-Cx26 protein could provide further insight into the role of this variant in hearing loss. With this objective, some functional studies were already performed by other groups.
Palmada et al. (2006) investigated the role of p.Arg127His mutation on hemichannel conductance in noncoupled oocytes. Western blotting ensured that p.Arg127His-Cx26 was inserted in the oocytes' membranes at a degree similar to wtCx26. In the subsequent measurements of hemichannel activity, p.Arg127His-Cx26 displayed a partially defective phenotype when expressed alone. Although coexpression of p.Arg127His-Cx26 with wtCx26 at equimolar levels (mimicking p.Arg127His heterozygosity) revealed no significant current difference relative to wtCx26 expressed alone, coexpression of p.Arg127His-Cx26 with wtCx30 or wtCx31 at equimolar levels (mimicking p.Arg127His homozygosity) revealed a strong dominant negative effect of the p.Arg127His-Cx26 mutant on each of the wild-type connexins.
Wang et al. (2003) performed dual whole-cell voltage-clamp assays in N2A cells stably transfected with cDNA encoding p.Arg127His-Cx26, and demonstrated that transjunctional current and macroscopic junctional conductance of p.Arg127His-Cx26 channels were greatly reduced. The authors also reported impaired neurobiotin transfer between pairs of N2A cells expressing p.Arg127His-Cx26 (n = 1 out of six pairs). These results seem not to be due to impaired translation or protein trafficking to the membrane, since Western blot revealed strong expression of the p.Arg127His-Cx26, and this mutant protein was predominantly expressed in the cell membrane being its immunocytochemical staining pattern indistinguishable from that of wtCx26. Bicego et al. (2006) and D'Andrea et al. (2002) also observed immunocalization of p.Arg127His-Cx26 similar to wtCx26, with junctional plaques in zones of cell-to-cell apposition, and Western blotting revealed that levels of p.Arg127His-Cx26 expression were comparable (small or no significant difference) with those of wtCx26. In these two studies, p.Arg127His-Cx26 channels have been found to have a much reduced permeability to Lucifer Yellow, but p.Arg127His-Cx26 did not show a dominant negative effect on wtCx26, regarding permeability to this tracer, when both variants were cotransfected at 1:1 ratio. p.Arg127His-Cx26 channels were also shown to fail diamidino-2-phenylindole (DAPI) transfer (D'Andrea et al., 2002).
Thönnissen et al. (2002) observed an elevated permeability of p.Arg127His-Cx26 channels to neurobiotin, contrary to the results obtained by Wang et al. (2003) above referred. It should, however, be noticed that in the study by Thönnissen et al. (2002), p.Arg127His-Cx26 was considerably more expressed than wtCx26; therefore, the high intercellular diffusion of neurobiotin observed with p.Arg127His-Cx26 channels (three times more than that obtained with wtCx26 channels) might not reflect the real permeability of each p.Arg127His-Cx26 channel; instead, it may be due to the very high number of p.Arg127His-Cx26 channels with reduced permeability to neurobiotin, as suggested by the results of Wang et al. (2003). This notoriously higher expression of p.Arg127His-Cx26 relative to wtCx26, which was not observed by D'Andrea et al. (2002) or by Bicego et al. (2006), might result from a different degree of purity of the constructs used for transfection, or it may have happened that the stably transfected p.Arg127His-Cx26 expressing clones chosen for subsequent analyses expressed, by chance, more Cx26 protein than the ones expressing wtCx26. It is also possible that the silent base change, from G to A, that the authors refer to exist at position 237 in the Cx26 sequence in all the constructs used might have altered protein expression levels in a manner that changed the relative expression of p.Arg127His-Cx26 and wtCx26.
In summary, the overall results clearly indicate an abnormal behavior of this mutation.
It is not known whether the alterations in the properties of p.Arg127His-Cx26 channels observed in vitro are meaningful at the biological level and, if so, at what extent or under which circumstances. If any in vivo effect exists, it might be potentiated or attenuated by other genetic and/or environmental factors. These still unknown interactions would explain the differences observed in the audiological phenotypes (normal or impaired) of the compound heterozygotes for p.Arg127His and p.Trp24X in family EL and in the family referred by Roux et al. (2004). This explanation could be extended to other cases of compound heterozygosity involving p.Arg127His and to the p.Arg127His homozygotes.
In the light of the genetic and functional data here analyzed, including the three Portuguese cases, we conclude that p.Arg127His mutation may in some cases be a cause of hearing loss, depending on environmental and/or genetic factors yet to be elucidated. Therefore, p.Arg127His should not be disregarded when providing genetic counseling to the families.
Footnotes
Acknowledgments
We would like to thank the family members for their collaboration. This study was supported by Fundação para a Ciência e a Tecnologia grants SFRH/BD/19988/2004 T.D.M. and SFRH/BD/24575/2005 (H.S.-T.).
Disclosure Statement
No competing financial interests exist.