
Abstract
Hereditary multiple exostoses (HME) is an autosomal dominant bone disorder characterized by growth of benign multiple exostoses. In our present study, we describe a four-generation Han Chinese kindred with eight members affected by HME. Haplotyping analysis and mutation detection was performed. The results linked the disease-causing gene to the EXT1 locus on chromosome 8. A novel mutation in EXT1, c.1897delC, which cosegregated with the disease phenotype, was detected. To further confirm this mutation, a mismatch primer was designed to introduce a ScaI restriction site into the normal allele by polymerase chain reaction, and the following restriction fragment length polymorphism analysis demonstrated that the mutation was not detected in any unaffected individuals of the family or 100 unrelated Han Chinese control individuals. This mutation leads to a frameshift from codon 633, resulting in a premature termination at codon 642 and loss of the highly conserved C terminal region of the protein. Therefore, this heterozygous mutation must be classified as pathogenic and can be regarded as the cause of HME in this Chinese family.
Introduction
H
Three genes are known to be associated with HME: EXT1 (MIM#133700), EXT2 (MIM#133701), and EXT3 (MIM#600209), located on 8q24.11-q24.13, 11p11-p12, and 19p, respectively. However, a second mutational hit may arise in a related gene such as the EXT-like genes or other genes involved in the signaling cascade of chondrocyte proliferation (Hall et al., 2002). The EXTL family of genes is related to EXT1 and EXT2 by sequence homology. The EXTL family of genes currently consists of three members: EXTL1, EXTL2, and EXTL3, respectively, located on 1p36.1, 1p11-p12, and 8p12-p22 (Wise et al., 1997; Wuyts et al., 1997; Van Hul et al., 1998). To date, no disorder has been attributed to mutations in EXT3, EXTL1, EXTL2, and EXTL3.
In this report, we describe a novel EXT1 mutation in one Han Chinese family with typical HME.
Materials and Methods
Patients and clinical study
A four-generation Han Chinese kindred in which HME had affected eight members (five men and three women; age range, 16-65 years) was investigated (Fig. 1). The diagnostic criteria for HME were radiological observations of at least two exostoses at the juxta-epiphyseal regions of the long bones (Legeai-Mallet et al., 1997b). The evaluation of the functional rating and severity were described previously (Francannet et al., 2001). Clinical photographs of five available affected individuals and radiographs from the proband were obtained. In addition, seven unaffected members (four men and three women; age range, 29-66 years) were included in the study. Blood samples were collected from the 12 family members (individual II-1, II-5, II-6, II-7, II-9, II-11, II-12, II-14, III-10, III-12, III-19, and III-20) by standard procedures with written informed consent.
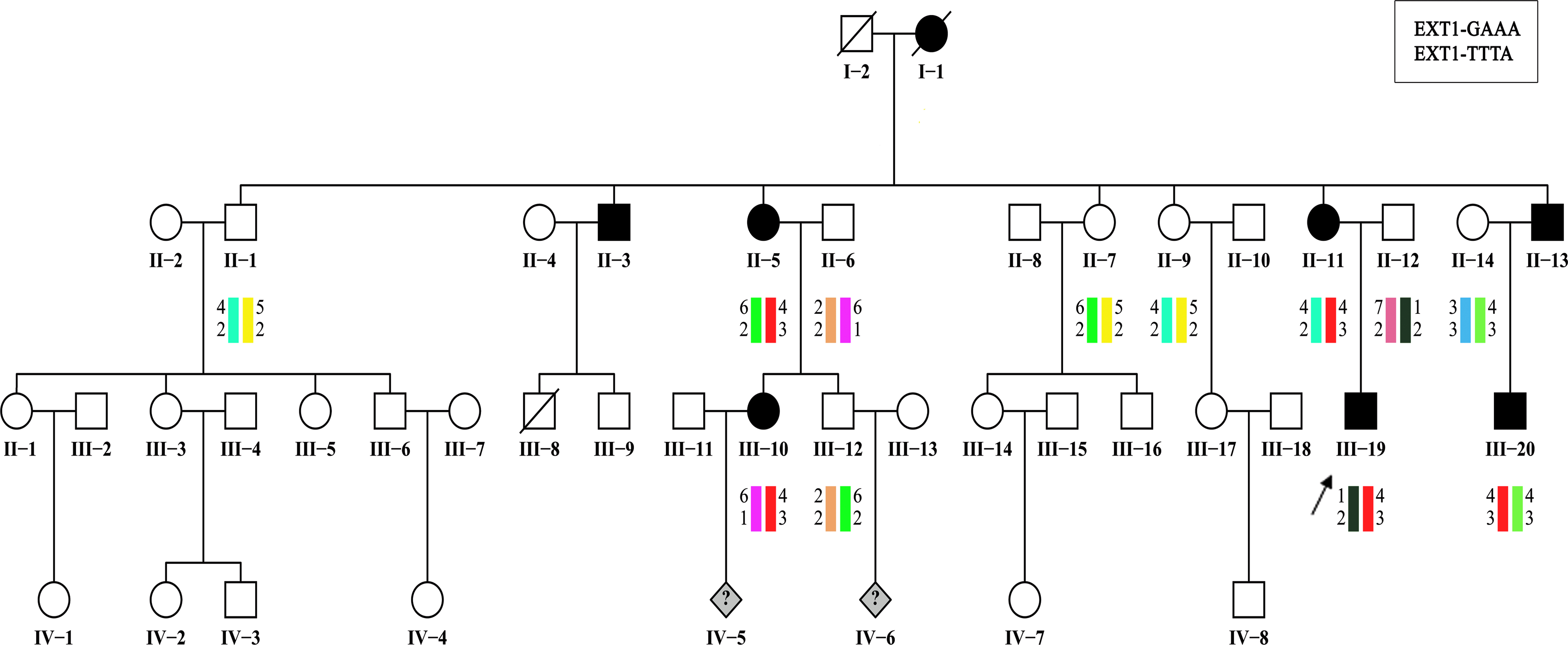
Pedigrees and disease-haplotype segregation of the Han Chinese family with HME. Filled symbols represent affected individuals with HME, and open symbols represent individuals without HME. Circles and squares indicate women and men, respectively. Rhombs indicate fetuses. The arrows identify the probands. The disease haplotype is red. HME, hereditary multiple exostoses.
Haplotyping analysis
Genomic DNA was extracted from venous blood samples using a proteinase K digestion and standard phenol-chloroform extraction. Using the UCSC Genome Browser on Human 2006 May assembly (www.genome.ucsc.edu), two perfect microsatellite repeat sequences each close to the EXT1 gene and the EXT2 gene were selected as genetic markers for haplotyping (Table 1). The design of the four primer pairs was based on the genomic sequences flanking the microsatellite repeats. Polymerase chain reaction (PCR) was performed under standard conditions. The PCR products were separated by electrophoresis on 8% denaturing polyacrylamide gel, and allele fragments were detected with routine silver staining.
F, forward primer; R, reverse primer.
Mutation analysis
All 11 exons of EXT1 were amplified by PCR using 13 pairs of primers (Table 2). PCRs were performed as follows: 2 × GC PCR buffer I, 125 U (5 U/μL) LA Taq DNA polymerase, 250 nM dNTPs, 10 μmol of each primer, and approximately 100 ng of template DNA in a total volume of 50 μL. PCRs were cycled in a 4 min 94°C predenaturation step followed by 35 cycles of a 30 s 94°C denaturation step, a 30 s 55.2°C-60.8°C annealing step, and a 30-45 s 72°C extension step depending on the fragment to be amplified. The PCR products were then subjected to automated DNA sequencing after purification.
The designation “ex” in each EXT1 primer name is followed by the exon number. The primers were designed from genomic sequences deposited in the UCSC Genome Browser on Human 2006 May assembly (www.genome.ucsc.edu).
F, forward primer; R, reverse primer.
Restriction fragment length polymorphism analysis of the mutation in EXT1
To further confirm this mutation, we employed a mismatch primer: 5′-ACCCATCTTTGATTTTTACAGATATTATCA
Results
Haplotyping analysis
Haplotype analysis indicated that the causative gene of the Han Chinese HME family was linked to EXT1 located on chromosome 8 (Fig. 1).
Mutation screening
We searched for pathogenic mutations in the proband of this family by direct sequencing of the PCR-amplified DNA fragments spanning all 11 exons of the EXT1 gene. We identified a heterozygous deletion in exon 10, c.1897delC (Fig. 2A). This deletion results in a frameshift from codon 633 and a premature termination at codon 642 and therefore designated as p.Leu633TyrfsX10 at the protein level. To confirm this deletion mutation, in all the other available affected family members, the exon 10 genomic fragment was subjected to automated DNA sequencing after purification. The results showed the cosegregation of mutation c.1897delC with the disease phenotype.

(
PCR-RFLP analysis
To further confirm this result, all the 12 available family members and 100 unrelated Han Chinese normal individuals were examined by PCR-RFLP analysis. Besides the normal 284 bp fragment, a 250 bp fragment was seen in all affected individuals from the family but was not detected in any unaffected family members or unrelated normal controls (Fig. 2B). Therefore, this heterozygous mutation must be classified as pathogenic and can be considered the causal mutation responsible for the phenotype of HME in this Chinese family.
Genotype-phenotype study
Five affected members from the family were physically examined. Details of the clinical information for the five available affected individuals are presented in Table 3. All these affected individuals had multiple exostoses that were located at the juxta-epiphyseal regions of long bones as well as other sites, such as tibia, humerus, radius, and ulna. All of them had mild short stature. Although affected individuals shared the same mutation, intrafamilial phenotypic variability was observed. The phenotype of individual III-20 was the most severe (Fig. 3), whereas other affected members exhibited moderate HME severity. Besides multiple exostoses that were located at the above sites, individual III-20 had abnormal bone remodeling leading to shortening and bowing of the forearms. The functional rating for the individual III-20 and other affected members is fair and good, respectively. No malignant transformation was observed in any of the affected individuals.
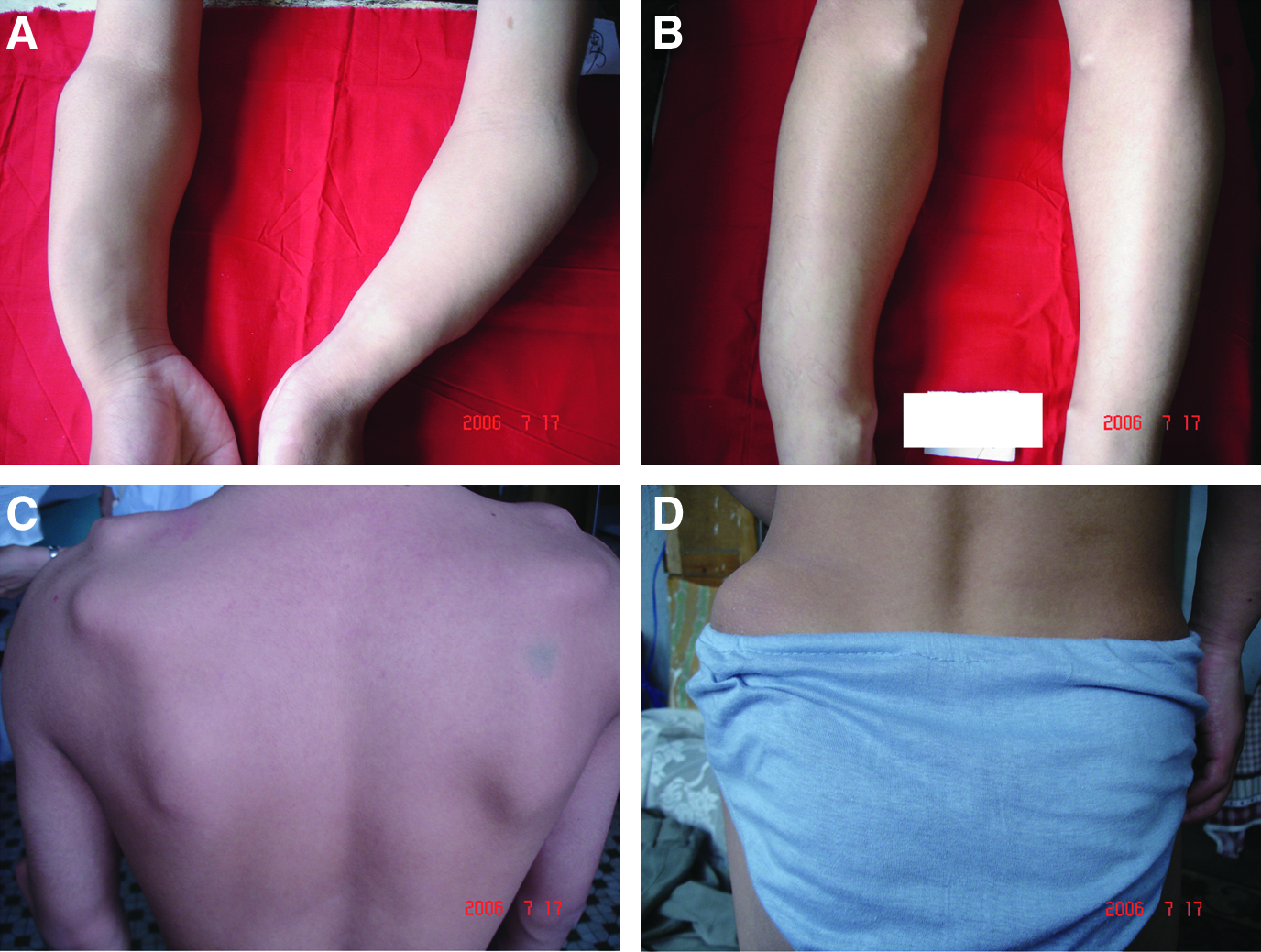
Photographs from individual III-20 with HME. Location of exostosis: (
Discussion
In this study, we investigated a Han Chinese family with typical HME. The disease locus was linked to the EXT1 gene and a new mutation (EXT1 c.1897delC) was identified. To determine whether the sequence variant identified in the EXT1 gene had previously been described, we checked in the Human Genome Mutation database (www.hgmd.cf.ac.uk/ac/index.php) and articles cited in the PubMed. We found there were no previous reports of this alteration.
The EXT1 and EXT2 genes are ubiquitously expressed at cell surfaces and in extracellular matrix. They encode proteins of 746 and 718 amino acids, respectively, which show homology to each other, especially at the carboxy terminus (Stickens et al., 1996; Wuyts et al., 1996). Both the EXT1 and EXT2 genes encode glycosyltransferases involved in heparan sulfate (HS) biosynthesis and probably have a tumor suppressor function (Raskind et al., 1995; Hecht et al., 1997). The novel EXT1 c.1897delC mutation causes a premature termination at codon 642, leading to deletion of the entire exon 11. This leads to the loss of the highly conserved C terminal region of the EXT1 protein, which appears to be involved in protein-protein interactions (Simmons et al., 1999). The truncated protein might lose its glycosyltransferase activity, which is critical for catalyzing the polymerization of HS (Philippe et al., 1997; McCormick et al., 2000; Francannet et al., 2001). HS chains interact with a variety of biological factors, such as growth factors, morphogens, and extracellular matrix proteins (Carey, 1997; Woods and Couchman, 1998); the biosynthesis of HS also regulates key events in embryonic development and homeostasis, and deranged HS biosynthesis could lead to diseases (Nadanaka and Kitagawa, 2008). Thus, this heterozygous mutation must be classified as pathogenic and it is likely to be of significance in the etiology of HME in this Chinese family, which is consistent with the autosomal dominant phenotype.
It has been reported that mutations in EXT1 and EXT2 are variable and random, without any mutation hotspots (Wuyts and Van Hul, 2000). According to Human Gene Mutation Database on HGMD Professional 2009.2 public total (www.hgmd.cf.ac.uk/ac/index.php), there are 144 mutations in EXT1 and 72 mutations in EXT2 that were identified in patients with HME. Mutation analysis of EXT1 and EXT2 genes has revealed that HME in most Caucasian patients are associated with EXT1 mutations (Philippe et al., 1997; Wells et al., 1997; Raskind et al., 1998; Wuyts et al., 1998). According to our current study and previous reports (Xu et al., 1999; Li et al., 2002, 2007; Shi et al., 2002; Hu et al., 2004; Chen et al., 2006; Xie et al., 2006; Liu et al., 2007, 2008; Zhao et al., 2009), EXT1 accounts for 26.4% (14/53) and EXT2 accounts for 39.6% of mutations (21/53) in Chinese patients with HME. These results also show that Chinese patients tend to be involved more frequently in EXT2 mutations than EXT1 mutations.
The previous genotype-phenotype studies suggested a strong correlation between EXT1 mutations and the severity of HME phenotypes, with respect to height, number of exostoses, and limited mobility (Francannet et al., 2001). However, in this study, intrafamilial phenotypic variability among affected members sharing the identical EXT1 mutation was observed; thus, the severity of HME is likely to be modulated by a differential genetic background and/or environmental factors. It could be speculated that mutations or polymorphisms in a modifier gene may modulate the disease phenotype. Further studies are still needed to elucidate a correlation between phenotype and genotype.
EXT1 mutations are more likely to trigger malignant degeneration of exostoses (Francannet et al., 2001; Porter et al., 2004). However, up till now, no instance of malignant transformation was seen among this EXT1-mutated family at the time of observation. This study therefore provides useful clinical information with respect to whether the lifetime risk of malignant degeneration of those affected individuals is higher than the normal individuals. Prenatal diagnosis for a fetus with high risk in this family can also be performed.
Footnotes
Acknowledgments
The authors would like to thank all the family members for their participation in the study. The authors are grateful to Dr. Supriya Srivastava from Cancer Science Institute of Singapore and Dr. Huijia Chen from A*STAR Institute of Medical Biology of Singapore for polishing this article. This work was supported by National Natural Science Foundation of China (No. 30971164) and Education Department of Liaoning Province (No. 20060908).
Disclosure Statement
The authors declare that there are no conflicts of interest.