
Abstract
Although few examples are formally documented, all polymerase chain reaction-based testing is theoretically vulnerable to allele drop-out (ADO), the failure to amplify one of the two alleles present in a cell. In a clinical setting, this can lead to false positive or negative diagnosis. We investigated the mechanisms leading to ADO in the MECP2 gene in two unrelated female patients undergoing testing for Rett syndrome. Both the patients had two benign DNA variations, c.819G > T and c.1161C > T, that appeared homozygous due to ADO. Bioinformatics analyses indicate that this region of the MECP2 gene is rich in complex tertiary structures called G-quadruplex and i-motifs, the disruption of which by the c.819G > T and c.1161C > T variants leads to preferential amplification of the variant allele. Other examples of ADO likely occur, and consideration of disrupting G-quadruplex and i-motif structures should be given when this phenomenon is unexpected. We identify factors in both the polymerase chain reaction amplification and the sequencing steps that help overcome ADO.
Introduction
A
Recently, Wenzel et al. (2009) described preferential allelic amplification in a patient appearing falsely homozygous for the c.400delT mutation in a G-C rich region of the MEN1 gene due to the formation of complex tertiary structures. In such G-C rich sequences, the G-rich strand can adopt a four-stranded G-quadruplex structure involving planar G-quartets stabilized by hydrogen bonding, whereas the complementary C-rich strand forms an i-motif, a tetrameric structure consisting of intercalated C-C+ base pairs (Burge et al., 2006; Guo et al., 2007). G-quadruplexes are highly prevalent in the human genome (Todd et al., 2005), and up to one-third are located within NCBI reference sequences. This suggests relevance to molecular diagnostic testing, given the potential effects of such structures on PCR efficiency (Wenzel et al., 2009).
One such molecular diagnostic test is for Rett syndrome (RTT; MIM no. 312750), a progressive neurological developmental disorder and the most common genetic cause of profound mental retardation in women, affecting 1:8000 girls (Hagberg et al., 1983; Laurvick et al., 2006). RTT was discovered to be caused by mutations in the X-linked MECP2 gene (Amir et al., 1999) and is now offered as a clinical test in many molecular genetics laboratories. Several groups have published methodologies for MECP2 mutation analysis and generally involve PCR and sequencing of the four coding exons (Amir et al., 1999; Wan et al., 1999), although some methods target common mutations.
Here we describe ADO in samples from two unrelated female patients displaying apparently homozygous variants, c.819G > T and c.1161C > T, in exon 4 of the MECP2 gene. This ADO was confirmed by allele-specific PCR, was not attributable to primer SNPs, and was not alleviated by the use of high fidelity polymerase in the amplification step. We show that the disruption of G-quadruplex and i-motif structures by these variants cause preferential amplification of the variant allele and identify factors in both the PCR amplification and the sequencing steps that alleviate this problem.
Materials and Methods
Patients
Samples were referred to the Children's Mercy Hospitals and Clinics Clinical Molecular Genetics Laboratory for clinical testing for RTT. Neither of the patients conformed to diagnostic criteria for classic RTT, but they displayed developmental delay, regression, and seizures. Parental samples were requested as part of routine follow-up due to apparent homozygosity. DNA was isolated from peripheral blood using a home brew salting out extraction procedure.
PCR procedure for amplifying exon 4 of MECP2
Standard
Our standard exon 4 PCR reaction (Fig. 1A; fragment A) is 1307-bp and is carried out in a 50 μL reaction volume containing 200 ng genomic DNA, 1 × PCR buffer (Tris-HCl [pH 8.8], 1.5 mM MgCl2, 0.5 mM KCl, 10 mM Tris; Stratagene, La Jolla, CA), 1 μmol dNTPs, 50 pmol each primer (1F: 5′ AGCCAGGCAGTGTGACTCTC and 6R: 5′ AAGCTTTGTCAGAGCCCTAC), and 0.25 μL Taq 2000 polymerase (Stratagene). PCR conditions are an initial denaturation at 95°C for 5 min, followed by 31 cycles of 30 s at 95°C, 30 s at 57°C, 1 min 30 s at 72°C, and a final extension step at 72°C for 10 min.

Amplification and sequencing strategy for exon 4 of the MECP2 gene with representative sequencing chromatograms. (
PfuTurbo DNA polymerase
This was used to amplify fragment A in a 50 μL reaction containing 100 ng genomic DNA, 1 × Pfu buffer (200 mM Tris-HCl [pH 8.8], 20 mM MgSO4, 100 mM KCl, 100 mM [NH4] 2SO4, 1% Triton X-100, 1 mg/mL nuclease-free bovine serum albumin), 25 pmol each primer (1F/6R), 1 μmol dNTPs, and 1 μL PfuTurbo Taq (Stratagene) using the standard PCR conditions described earlier.
Alternative primers used for amplification
Fragment B (Fig. 1A) is a 609 internal fragment generated with primers 2F: 5′ AAGTCCTGGGAAGCTCCTTG and 5R: 5′ ATCTTCTCCTCTTTGCAGAC using otherwise standard reaction conditions. An upstream primer, MECP2-165:5′ TGAGTGGCTTTGGTGACAGG was used with primer 6R. Fragment C was generated using primers 2F: 5′ AAGTCCTGGGAAGCTCCTTG and 3R: 5′ GGCTTTCTTTTTGGCCTCGG. Fragment D was generated using primers 4F: 5′ ACTGAAGACCTGTAAGAGCC and 5R: 5′ ATCTTCTCCTCTTTGCAGAC.
DNA sequencing and analysis
PCR products were purified using Exo-SAP-IT (USB Corporation, Cleveland, OH) according to the manufacturer's instructions. Both the forward and reverse strands of the purified PCR product were sequenced using fluorescent dye-terminator sequencing (BigDye; Applied Biosystems, Foster City, CA),. Sequencing reactions were purified using spin columns (Princeton Separations, Adelphia, NJ) or the BigDye XTerminator Purification Kit (Applied Biosystems) according to the manufacturer's instructions. Results were analyzed on either an ABI 310 or ABI 3130 genetic analyzer (Applied Biosystems). Sequence results were compared with published reference sequence (NM_014795.2) using Sequencher 4.5 (Gene Codes Corporation, Ann Arbor, MI). For MECP2 deletion analysis, multiple ligation probe assay (MLPA) (Kit no. P015-MECP2, MRC Holland, Amsterdam, The Netherlands) was performed according to the manufacturer's instructions.
Allele-specific PCR
To demonstrate that we were experiencing ADO, we designed allele-specific primer sets for both polymorphisms at the c.819 and c.1161 sites. For c.819G/T, our MECP2-2F primer (as described earlier) was used in combination with either reverse primer, 819W: 5′ GCGGCTGCCACCACACTC or 819M: 5′ GCGGCTGCCACCACACTA to generate a 194-bp product. For c.1161C/T, 4F (as described earlier) was used in combination with either reverse primer, 1161W: 5′ GGCTCAGGTGGAGGTGGG or 1161M: 5′ GGCTCAGGTGGAGGTGGA to generate a 173-bp product. For all reactions, an additional primer set targeting the factor V gene was used in the same tube as an amplification control, generating a 223-bp product. A standard touchdown PCR protocol with an initial 64°C annealing step was used.
Bioinformatics
G-quadruplex and i-motif-forming sequences were identified by running two web servers: QGRS Mapper (http://bioinformatics.ramapo.edu/QGRS/) (Kikin et al., 2006) and Quadfinder (http://miracle.igib.res.in/quadfinder/) (Scaria et al., 2006). Neither of the web servers checks the antisense strand of the input sequence, so the reverse complement must also be checked. The pattern input for G-quadruplex is formed by four core sub-motifs, each being 2-5 “Gs” separated by 1-7 unspecified nucleotides (N) called loop length, that is, G2-5N1-7G2-5N1-7G2-5N1-7G2-5. Similarly, the search pattern for i-motifs used was C2-5N1-7C2-5N1-7C2-5N1-7C2-5. If a G-quartet is found in a sequence, then an i-motif is on the same location of its reverse strand or vice versa. Only nonoverlapping motifs are reported.
Results
We performed our standard PCR and sequencing of the MECP2 gene on two unrelated female patients received in our clinical laboratory. Although the samples were negative for pathogenic mutations, both had two known benign polymorphisms, c.819G > T (p.G273G) and c.1161C > T (p.P387P), in exon 4. Although the sequence obtained was clear, the chromatogram at nucleotide c.819 was ambiguous due to minor shouldering that could not be resolved upon numerous repeats using multiple sequencing primers; we were unable to determine whether it was homozygous or heterozygous. However, the c.1161 appeared homozygous T/T in both forward and reverse reactions, despite numerous repeats (Fig. 1B; first column). Although we had previously not seen these variants in our laboratory, both are listed in the Rett syndrome database RettBASE: www.mecp2.chw.edu.au/with 5 entries for c.819G > T, one for c.1161C > T, and one joint entry. Regardless, they are sufficiently rare to be suspicious of homozygosity in the absence of consanguinity. This finding was confirmed by a second clinical molecular genetics laboratory.
Polymorphisms within the binding sites of the primers were ruled out as possible causes of ADO by using an alternative set of internal primers that cover both variants in a 609-bp fragment and saw no difference in the chromatogram (Fig. 1; primer set B). We also designed a forward primer further upstream to sequence through the primer binding region in our standard reaction; neither patient had an SNP. Despite the ambiguous data for the c.819 variant, we considered the possibility that both variants were actually homozygous due to a deletion on the opposite allele. However, MLPA results indicate that both patients are negative for MECP2 deletions (data not shown). Parental testing was done to investigate the possibility that each parent carries both variants and that the patients are truly homozygous. We performed our standard PCR and sequencing of exon 4 and were surprised to find that the mothers of both patients have results identical to their daughters: ambiguous results for c.819 and homozygous T/T for c.1161 (data not shown). One of the fathers was tested and found to be negative for both variants; the father of the second patient was unavailable. The third solution we explored was the possibility of maternal uniparental disomy for the X chromosome. Using the polymorphic CGG repeat in the FMR1 gene to show distinct patterns for patient 1 and both her parents, we were able to verify the presence of a paternal allele (data not shown). While this finding rules out whole chromosome uniparental disomy (UPD) for the X chromosome, we acknowledge that segmental UPD was a theoretical possibility that we opted not to explore since, in the absence of multiple unlikely events, it would not explain why the mothers also appear homozygous.
Allele-specific PCR to demonstrate ADO
To demonstrate that ADO is responsible for the apparent false homozygosity, we designed allele-specific primers for wildtype and mutant alleles for both variants. As shown in Figure 2, patient 1 and her mother are heterozygous for both variants and the father is negative, thus proving that we were indeed experiencing ADO.
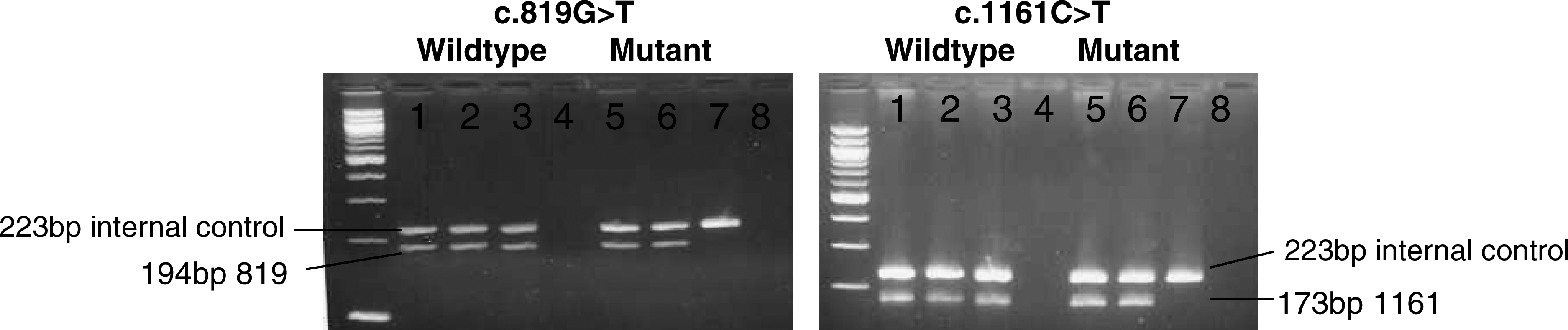
Allele-specific amplification of the c.819G > T and c.1161C > T variants. For each, lanes 1-4 are allele-specific reactions for the wildtype sequence; lanes 5-8 are designed for the mutant. The c.819 reaction yields a product of 194-bp; for c.1161, the amplicon is 173-bp. The internal control is a 223-bp band generated by primers targeting the factor V gene. Lanes 1 and 5 show the heterozygous patient; lanes 2 and 6 show the heterozygous mother; and lanes 3 and 7 show the wildtype (hemizygous) father. Lanes 4 and 8 are no template control.
Sequence structure as a cause of ADO
Having eliminated genetic causes for our sequencing results as well as primer SNPs causing ADO, we next examined the local sequence structure for elements that might contribute to ADO. Our standard 1307-bp exon 4 PCR product is GC-rich (∼53%) with the dinucleotides GG and CC numbering 109 and 156 counts, respectively. Such sequences potentially harbor G-quadruplexes and i-motifs that could interfere with PCR amplification. Using QGRS Mapper (http://bioinformatics.ramapo.edu/QGRS/) and Quadfinder (http://miracle.igib.res.in/quadfinder/), we searched for G-quadruplex patterns (G2-5N1-7G2-5N1-7G2-5N1-7G2-5) and i-motifs (C2-5N1-7C2-5N1-7C2-5N1-7C2-5) in this amplicon and found 10 nonoverlapping G-quartets in addition to 13 nonoverlapping i-motifs (see Fig. 3). Since this sequence is so rich in G-quartets and i-motifs (they are complementary on double strands), we concluded that stable tertiary structures were likely contributing to ADO in our samples. In particular, the T variant at c.819 replaces the third G in a G-quadruplex sequence (

Amplified region of MECP2 exon 4. Amplification and sequencing primers used (shown in bold with primer numbered with superscript), overall distribution of i-motifs (broken underline), and G-quadruplexes (double underline). Polymorphisms occurring on i-motifs and G-quadruplex sequences are highlighted; the c.819 and c.1161 positions are in bold. Flanking intronic sequences are gray.
Resolving ADO
PCR conditions
In an attempt to alleviate ADO in our samples, we first tried PfuTurbo Taq in the amplification step, because a high fidelity Taq has been reported to resolve amplification bias in the CAH gene (Schulze et al., 1998). Surprisingly, we saw little improvement in the sequencing chromatogram (Fig. 1B; second column). Another factor previously reported to affect the amplification ratio is magnesium concentration (Wenzel et al., 2009), so we tried increasing our standard 1.5 mM final concentration to 2 and 3.2 mM. The sequencing chromatogram derived from the 2 mM product was actually slightly more biased toward the mutant strand compared with the standard conditions; the 3.2 mM product failed to amplify. Other additives to the standard reaction that failed to improve or resolve ADO include 5% dimethyl sulfoxide (DMSO) or 2 mM betaine (Fig. 1B; columns 4 and 6). One PCR condition that did show improved results was raising the annealing temperature of the otherwise standard PCR product to 64°C: It completely resolved the bias for c.819 but showed little improvement for c.1161 (Fig. 1B, column 5).
Since structural motifs appear to be causative for ADO, we hypothesized that separately amplifying each variant (Fig. 1A; fragments C and D) could solve the problem, as this would be expected to dramatically alter the formation of tertiary structures. This approach was able to completely alleviate ADO for the c.819 variant but showed little improvement for the c.1161 variant (Fig. 1B, columns 8 and 9), which is actually embedded in a strong i-motif sequence. In this case, the wildtype allele is likely more refractory to amplification, as a “T” in that position would destabilize the structure, resulting in preference for the variant strand. Regardless of the improvements we saw by using alternative PCR primers, we wanted a solution that could be applied to our existing procedure; thus, subsequent experiments were performed using the standard primer set (fragment A) amplified and using a 64°C annealing temperature.
Sequencing conditions
While doing the experiments just described, we also noticed differences in chromatograms generated from the same PCR product using different sequencing primers. This could explain why ADO for only one variant (c.819) was resolved by raising the PCR annealing temperature to 64°C, despite presumed coamplification of the two variants on the same strand. This observation prompted us to experiment with sequencing conditions to see whether further resolution of the amplification bias could be obtained. Since the PCR product generated at 64°C annealing had yielded the best results, we used that as the template for sequencing reactions containing no additives, 5% DMSO, or 2 M betaine in the reaction; all were run at an annealing temperature at 58°C versus the standard 55°C for the sequencing reaction. A higher annealing temperature for the sequencing step led to some degree of improvement for all three reactions (no additive, 5% DMSO, and 2 M betaine); however, the 2 mM betaine had the highest percentage of wildtype peak for c.1161, though considerable bias toward the mutant peak still remained (Fig. 1C). In addition, there was a considerable difference in the chromatograms from otherwise identical sequencing reactions when using primer 2 versus primer 4 as the sequencing primer, with primer 2 showing much more of the wildtype peak than the chromatogram from primer 4. As expected, the c.819 variant appeared heterozygous, as it did while using standard sequencing temperatures. Finally, we sequenced a template amplified with PfuTurbo Taq under standard conditions (64°C annealing failed to amplify), using 2 mM betaine/58°C annealing for the sequencing reaction, and this proved to be the best combination for both variants. While there is still bias toward the mutant allele, the chromatogram is clear enough to be called heterozygous.
Discussion
Our data show that G-quadruplex and i-motif sequences cause reproducible effects on amplification efficiency as well as the sequencing reaction, contributing to ADO in the MECP2 gene. The formation of G-quadruplex or i-motif secondary structures makes PCR amplification difficult, whereas destabilization of such structures ameliorates such situations. For the c.891G > T allele, the wild type has the “G” on a G-quartet. However, this G-quadruplex structure is not very strong, as there is no loop (i.e., no other nucleotides inserted) between the first two segments “GG-GG.” The G > T variation completely destabilizes this G-quadruplex structure, resulting in preferential amplification of the mutant strand. The “C” at position c.1161 falls within a strong i-motif secondary structure on the sense strand and, therefore, a G-quadruplex on the antisense strand. When mutated to “T,” these structures are weakened, though still established, and result in preferential amplification of the mutant strand in the PCR reaction.
The c.819G > T and c.1161C > T polymorphisms have been reported by two separate groups in the literature, neither with mention of amplification bias (Lesca et al., 2007; Zahorakova et al., 2007). Zahorakova et al. (2007) detected both variants in the heterozygous state in the same patient (personal communication), although in the absence of parental testing it is not known whether the variants are on the same allele, as in our patients. Examining the reaction conditions used by Zahorakova et al. (2007) explains why ADO was not a problem: They used primers that amplified each variant separately, an annealing temperature of 63°C, and a very different reaction buffer (150 mM Tris-HCl, pH 8.8, 40 mM (NH4) 2SO4, 0.02% Tween 20, 5 mM MgCl2, Top-Bio, Prague, Czech Republic), all of which could potentially contribute to more efficient amplification. Lesca et al. (2007) reported the c.819G > T variant alone; their choice of primers explains why ADO was not a problem, as they used a primer set comparable with our primer set 2-4, which was sufficient to avoid ADO.
Our work indicates that optimization of both the PCR and sequencing steps is important for avoiding ADO. We obtained the least amount of ADO in our samples by using a high fidelity polymerase for the amplification step and sequencing with a 58°C annealing temperature and 2 mM betaine in the reaction. Betaine reduces the formation of secondary structure in GC-rich regions and eliminates the base pair composition dependence of DNA melting, so it may help to lower annealing temperature requirements and destabilize G-quadruplex and i-motif structures (Rees et al., 1993). Raising the annealing temperature of the PCR reaction to 64°C also showed marked improvement in the chromatogram, although slightly less than using Pfu Taq. The G-rich strand may adopt a quadruplex conformation involving G-quartets, whereas the C-rich antisense strand likely folds into i-motifs based on intercalated C•C+ base pairs. Obviously, both G- and C-rich complementary strands may freely exist or form Watson-Crick base pairing (i.e., duplex conformation). The distribution of different molecular conformations in solution depends on reaction conditions such as pH, temperature, and salt concentrations. In a PCR reaction, the G-quadruplex and the i-motif would efficiently compete with the other conformations and become dominant in the molecular population in high temperatures or low pH (Phan and Mergny, 2002), whereas the free single-stranded DNA molecules are relatively rare. However, the tertiary structures in the G- and/or C-rich strands could be largely destabilized through elevated temperatures or genetic mutation. This explains why the mutant types predominate in the experiments and ADO could be ameliorated by higher annealing temperatures in the PCR (e.g., 64°C) and the sequencing steps.
In addition to the report by Wenzel et al. (2009), there is one other description of ADO in the absence of primer SNPs in the literature: Day et al. (1996) reported false homozygosity for the common 21-hydroxylase gene mutation, conversion of A or C at nt656 to G in intron 2. It was later demonstrated that this problem could be alleviated by using a high fidelity Taq/Pwo polymerase in the PCR step (Schulze et al., 1998). We analyzed the CYP21 gene and found it to be rich in G-quadruplex and i-motifs, which are the likely cause of ADO in this gene, presumably because the wildtype “C” has a stronger binding structure than the mutant “G” type.
Though our work is specific to two variants in the MECP2 gene, it has obvious implications for other MECP2 variants as well as PCR-based analysis of any other gene, particularly when amplifying G-C rich sequence. Twelve polymorphisms listed in RettBase occur in G-quadruplex sequences, and 12 others occur in i-motif sequences (Fig. 3). Two polymorphsims present in i-motif sequences, c.1126C > T and c.1335C > A, have been detected in our laboratory with no evidence of ADO or amplification bias. However, these variants are not predicted to disrupt the tertiary structures (i.e., G-quartets or i-motifs) formed in the wild type, so both wildtype and mutant molecule species are equally represented in the reaction, and ADO was not a problem. Caution should be used in primer design for any PCR-based assay; the target sequence should be checked for potential structures disruptive to amplification. In addition, optimization of the reaction conditions and careful validation are necessary to avoid misdiagnoses in clinical settings.
Footnotes
Acknowledgment
We gratefully acknowledge the helpful comments of Dr. Shihui Yu.
Disclosure Statement
No competing financial interests exist.