
Abstract
Aim: The aim of this work was a haplotype analysis of the major mutations (C282Y, H63D, S65C) and IVS2(+4)t/c, IVS4(−44)t/c, and IVS5(−47)a/g polymorphisms of the hemochromatosis HFE gene in populations inhabiting the territories of Russia (Russians, Finno-Ugrians, Central Asians, and Arctic Mongoloids). Method: The hemochromatosis gene (HFE) alleles were detected using the polymerase chain reaction/restriction fragment length polymorphism method. Results: Of the eight possible intronic haplotype variants, the TTG, TTA, CTA, and CCA were identified. The HFE alleles with the different haplotype variants were distributed in an ethnospecific manner among the populations. Our finding was that every one of the C282Y, H63D, and S65C mutations was in linkage disequilibrium only with one of the intronic haplotype variants: TTG, CTA, and CCA, respectively. The data from context analysis of DNA regions where the examined single-nucleotide polymorphisms are located suggested their involvement in splicing. Conclusions: Different genotypes of the HFE gene occur at different frequencies among populations of Russia. Carriers of the specific genotype variants may potentially express distinct sets of alternative HFE mRNAs.
Introduction
T
HFE protein regulates transferrin receptor 1 (TfR1)-dependent iron uptake in many cell types (Feder et al., 1996; Gross et al., 1998; Lebron et al., 1998; Lieu et al., 2001; Waheed et al., 2002). HFE interacts with transferrin receptor 2 (TfR2) (Griffiths and Cox, 2003; Goswami and Andrews, 2006) expressed in a smaller number of cells than TfR1 (Vogt et al., 2003). HFE is required for normal regulation of hepcidin synthesis in liver and hepcidin-mediated iron export from macrophages, enterocytes, and hepatocytes (Nicolas et al., 2001; Ahmad et al., 2002; Bridle et al., 2003; Makui et al., 2005).
The C282Y polymorphism in the HFE has accrued interest to this gene. A cystein-to-tyrosine substitution at residue 282 in the α3 protein domain interferes with the HFE-β2m interaction, thereby hindering the passage of the protein from endoplasmic reticulum to cell surface (Feder et al., 1996, 1997; Waheed et al., 1997). A high percentage of patients with hereditary hemochromatosis type 1 (HH1) (the estimate varies from 50% to 100%, depending on ethnic reference) are homozygotes for the C282Y mutation (Beutler et al., 1996; Feder et al., 1996; Merryweather-Clarke et al., 2000; Hanson et al., 2001). A reduced frequency of the C282Y/C282Y homozygotes was observed among HH patients in Russia (Mikhailova et al., 2006; Potekhina et al., 2005). They suffer from excess of iron in parenchymal cells and its deficiency in reticuloendothelial cells and enterocytes (Lieu et al., 2001; Drakesmith et al., 2002; Pietrangelo, 2003). Clinical penetrance of HH1 among the C282Y homozygotes in human populations is incomplete, varying widely from 0.65% to 29% (Jackson et al., 2001; Beutler et al., 2002; McCune et al., 2002; Ajioka and Kushner, 2003). Other genetic and environmental factors contribute to the formation of the HH1 phenotype (Beutler, 2003). H63D is another major missense mutation associated with iron overload, yet with its mild forms (Feder et al., 1996; Beutler, 1997; Mura et al., 1999). C282Y/H63D compound heterozygotes occur among HH1 patients at frequencies varying from 0% to 10% (Beutler et al., 1996; Beutler, 1997; Mura et al., 1999; Merryweather-Clarke et al., 2000; Holmström et al., 2002). Unlike C282Y, the H63D mutation does not disrupt the interaction of HFE with β2m, has no effect on the binding of HFE to TfR1, but inhibits the ability of HFE to regulate iron export from macrophages (Feder et al., 1997; Waheed et al., 1997; Montosi et al., 2000; Drakesmith et al., 2002; Davies and Enns, 2004). There are data indicating that the less widespread S65C mutation is also associated with mild forms of iron overload, especially in compound heterozygotes with the C282Y or H63D alleles (Mura et al., 1999; Holmström et al., 2002).
Cell type-specific features of HFE interaction with the regulators of iron homeostasis, such as TfR1 (Waheed et al., 2002), TfR2 (Goswami and Andrews, 2006), and hepcidin (Bridle et al., 2003; Ludwiczek et al., 2004; Fleming and Britton, 2006), are thought provoking. More must be known about the mechanisms that regulate the expression of the HFE gene itself in different cell types (Davies and Enns, 2004). Spatial localization of the HFE protein is varied among cell types (Parkkila et al., 1997a, 1997b; Zhang et al., 2004; Bastin et al., 2006). Like the pre-mRNAs of the other nonclassical MHC I molecules (Fujii et al., 1994), the HFE transcripts are subjected to alternative splicing. The number of mRNA isoforms found is considerable, but their observed spectrum varies widely (Jeffrey et al., 1999; Rhodes and Trowsdale, 1999; Thenie et al., 2000; Sánchez et al., 2001). One reason may be the variability in the nucleotide sequences of the regions involved in HFE pre-mRNA splicing. The HFE gene is quite polymorphic, and the composition of the intragenic haplotypes varies among races and probably ethnic groups (Toomajian and Kreitman, 2002). A part of the identified polymorphic sites may be relevant to splicing routine (De Villers et al., 1999; Steiner et al., 2002; Floreani et al., 2005), that is, they may have an effect on the diversity of the HFE gene products.
In this study, we analyzed the distribution of the major mutations (C282Y, H63D, and S65C) in the HFE gene and intragenic haplotypes defined by intronic polymorphisms [IVS2(+4)t/c, IVS4(−44)t/c, IVS5(−47)a/g] in Asian and Caucasoid ethnic groups in territories of Russia. The distribution of the four identified haplotypes—TTG, TTA, CTA, and CCA—for these polymorphic sites of the HFE gene is ethnospecific in the human populations. We inferred that each of the C282Y, H63D, and S65C mutations was in disequilibrium with a distinct intronic haplotype: TTG, CTA, and CCA, respectively. On the basis of DNA context analysis, we showed that the given intronic polymorphisms may be potentially relevant to splicing.
Materials and Methods
Sampling
The Russian sample was drawn from the inhabitants of the Novosibirsk city. The pooled sample of the Finno-Ugric group included representatives of the Mordovian, Khanty, and Mansi peoples inhabiting along the basin of the Ob’ river. Other samples were drawn from Tuvinians, the inhabitants of the Altai-Sayan plateau (capital Kyzil), and from Altaians and Kazakhs (the Kosh-Agach region, the Altai republic), which are referred to the Central Asian peoples. Samples were also obtained from Nivkhi, the Eastern Asian people inhabiting along the basin of the Amur River and in the Sakhalin Island, and also from Chukchees, the Asians inhabiting settlements of the Chukchee Autonomous Area.
DNA extraction
Genomic DNA was extracted from peripheral blood leucocytes by the standard phenol-chloroform method (Sambrook et al., 1989).
DNA amplification, restriction, and electrophoresis
The nucleotide sequence of the human HFE gene from GenBank (accession no. Z92910) was used for primer choice. Conditions for amplification and restriction fragment length polymorphism analysis are given in Supplemental Table S1 (available online at www.liebertonline.com).
PCR reaction mixtures consisted of 0.5-1 μg of genomic DNA, direct and reverse primers at 0.4 μM, 0.1 mM of each of the deoxynucleotide triphosphates, 1.5 mM MgCl2, 10% dimethylsulfoxide, 0.01% Tween-20, 20 mM (NH4)2SO4, 75 mM Tris-HCl (pH 9.0), and 1.25 U Taq DNA-polymerase. The total volume of the reaction mixture was 25 μL. PCR was performed using an Eppendorf Mastercycler gradient amplifier (Eppendorf Scientific). The PCR products were digested with a restriction endonuclease (Supplemental Table S1), and the resulting fragments were electrophoresed on a 5% polyacrylamide gel.
Linkage analysis between the C282Y, H63D, and S65C mutations and haplotypes at the IVS2(+4), IVS4(−44), and IVS5(−47) polymorphic sites in the HFE gene
The samples from various ethnic groups described earlier and from a group of patients with chronic multifactorial diseases (Mikhailova et al., 2006) were used for linkage analysis. Samples were drawn from all the TTG/TTG and CTA/CTA homozygotes to analyze the linkages to the C282Y and H63D mutations, respectively. As regards the S65C mutation, the S65C/wild type (WT) genotype carriers with the CCA/CCA alleles and the H63D/S65C compound heterozygotes were included in the analysis. Chi-square contingency table analysis was performed to determine linkage disequilibrium (LD) between the mutations and the intronic haplotype.
Haplotype analysis of the ethnic groups
The intragenic haplotypes for the IVS2(+4)t/c, IVS4(−44)t/c, and IVS5(−47)a/g polymorphisms were determined in the HFE gene sequences in the samples from various ethnic groups. The cohorts used for the estimation of exonic and intronic polymorphisms' frequencies differed in size, and therefore, the haplotype analysis data in the ethnic groups were recalculated to obtain the correct estimates. The TTG, CTA, TTA, and CCA haplotype frequencies were estimated in WT chromosomes only, and then the results were calculated according to the WT frequency, defined earlier in all the ethnic groups examined. Population haplotype frequencies were weighted by fw, where f is the frequency of a concrete intronic haplotype on the WT chromosomes of the particular population and w is the weight of the WT alleles in this population.
Derivation of the minimum parsimony tree from data on the HFE gene polymorphisms
We invoked auxiliary data on haplotype diversity at the HFE locus for the three major continental human populations (Toomajian and Kreitman, 2002) (Supplemental Table S2, available online at www.liebertonline.com) to assess the relationships between alleles with the different intronic haplotypes. The Dollo parsimony method was applied to these data using the DOLLOP program of the PHYLIP package (Felsenstein, 1989) with the default parameters. The target tree from the total 13 with the minimum 15 reversions was selected as the one with the total minimum number of reversions omitting the rearranged set of related positions 279, 782, 1436, and 2469.
Results
Genotype data on the C282Y, H63D, and S65C mutations in the HFE gene in Siberian populations are summarized in Table 1.
Size of the sample typed for the C282Y and H63D alleles only; number in parentheses indicates the size of the sample typed for the S65C allele. WT are gene variants without the C282Y, H63D, and S65C mutations.
The frequencies of each of the three mutations in the HFE gene in Russians were close to those established for many Central European ethnic groups (Merryweather-Clarke et al., 2000). The C282Y mutation was detected at a frequency of 3.6% in Russians and also at low frequency in the Tuvinian and Chukchee populations, presumably because of Caucasian admixture. Cases of HH for representatives of the Asian ethnic groups in Russia have not been described in the literature. H63D was much more widespread. Mutation frequencies close to those in Russians were found in the Finno-Ugric group. In the Asian ethnic groups, the H63D frequencies decreased eastward, being lowest in the Chukchees. The S65C mutation was found at low frequencies (1.2%-1.7%) in Russians, Chukchees, and Mansis.
The DNA samples selected in the course of the genotype analysis were pooled together with DNA samples from Russians affected with various chronic diseases. The total sample was subjected to haplotype analysis for the three intronic polymorphisms, IVS2(+4)t/c, IVS4(−44)t/c, and IVS5(−47)a/g in the HFE gene. The results are given in Table 2.
The most probable haplotypes are shown in italics; unambiguously identified genotypes used in linkage disequilibrium analysis are marked in gray.
The genotypes marked in gray (Table 2) were unambiguously defined and were used for LD estimations between the C282Y, H63D, and S65C mutations and haplotypes at the IVS2(+4), IVS4(−44), and IVS5(−47) sites. The C282Y mutation was in LD with TTG intronic haplotype [χ2 (df = 1) = 76.0; p = 2.7 × 10−18], the H63D with CTA [χ2 (df = 1) = 271.8; p = 4.7 × 10−61], and the S65C with CCA [χ2 (df = 1) = 26.8; p = 2.3 × 10−7]. All the other identified genotypes were consistent with the above inferences.
In all the ethnic groups examined, we identified among unambiguous cases only four variants of haplotypes, TTG, CTA, CCA, and TTA (Table 2). Taken together, the population data in Tables 1 and 2 allowed us to estimate the frequencies of the haplotype variants in the gene pools of the geographically distant ethnic groups of Siberia. The observed and calculated values are given in Table 3.
The TTG haplotype was widespread in all the Siberian ethnic groups. High frequency of the CCA haplotype in WT chromosomes is a characteristic feature of all the Asian groups, being much lower among the Russians and Finno-Ugrics (Table 3). About every 7th Russian and 24th Chukchee bearer of that haplotype had the S65C mutation. The S65C mutation was not found in the Central Asian populations (Tuvinians, Altaians, and Kazakhs), although the CCA haplotype frequency was very high in these ethnic groups. The highest percentage, 25%, of the CTA variant was for Russians, and its frequency was lower for Finno-Ugrics and Central Asians. CTA haplotypes identified in Chukchees occurred only on chromosomes carrying the H63D mutation. In terms of the TTA haplotype frequency, Russians were close to Kazakhs and Tuvinians.
Thus, the distribution of the genotypes for intronic haplotypes of the HFE gene was ethnospecific, and the Arctic Asians (Chukchees) differed clearly from the Central Asian groups in this respect.
Discussion
Haplotype variants for the IVS2(+4)t/c, IVS4(−44)t/c, and IVS5(−47)g/a polymorphisms in the HFE gene: Association of each of the C282Y, H63D, and S65C mutations with a specific intronic haplotype variant
Pooled DNA samples from C282Y, H63D, and S65C carriers selected from ethnospecific groups throughout Russia and from patients with various pathological conditions were typed for three intronic polymorphic sites in the HFE gene and the structure of the haplotypes was determined. Of the eight possible HFE gene intronic haplotype variants, we unambiguously identified TTG, TTA, CTA, and CCA only in various ethnic backgrounds (Table 2). Our results are consistent with those previously obtained for the three continental human populations (Toomajian and Kreitman, 2002). However, the presence of the CTG, TCA, and CCG variants in addition to those listed above has been revealed (Beutler and West, 1997; Rochette et al., 1999). We confirmed the linkage for C282Y with the TTG haplotype and for H63D with the CTA variant (Beutler and West, 1997; Toomajian and Kreitman, 2002; De Lucas et al., 2005). Our novel finding was the association between the S65C mutation and the CCA haplotype (Table 2).
Ethnospecific distribution of the intronic haplotype variants of the HFE gene in populations of Russia
The observed and calculated frequencies of intronic haplotypes associated with mutations or without them in geographically distinct human populations of Russia are presented in Table 3. Four variants of the haplotype set for three analyzed intronic polymorphisms in the HFE gene were found in every ethnic group. We established the highest level of the HFE polymorphism for Russians. The TTG haplotype was widely spread in all the populations of northern Asia, but in different proportions. It was prevalent among the Finno-Ugric and occurred at high frequencies in Russians and Chukchees (Table 3). CCA occurred at a frequency of 41%-55% among Asians and at lower frequencies among Finno-Ugrics and Russians. The CTA haplotype was virtually absent in Chukchis, with highest frequency in Russians and Finno-Ugrics. The TTA haplotype occurred at close frequencies in groups of Russians, Kazakhs, and Tuvinians (13%-17%) and at lower frequencies in those of Finno-Ugrics and Chukchees (Table 3).
The HFE polymorphism features
According to the data of Toomajian and Kreitman (2002), each of the two structurally most heterogenous TTG and CTA allele classes can be divided into three sequence groups depending on single-nucleotide polymorphism (SNP) pattern specificity. The TTA alleles belong obviously to one sequence group with a common ancestor. There is just a single case of CCA allele variant (Supplemental Fig. S1, available online at www.liebertonline.com).
Figure 1 gives an overall pattern of the sequential accumulation of mutations in the HFE gene—the emergence order of allele variants with the TTA, TTG, CCA, and CTA haplotypes.
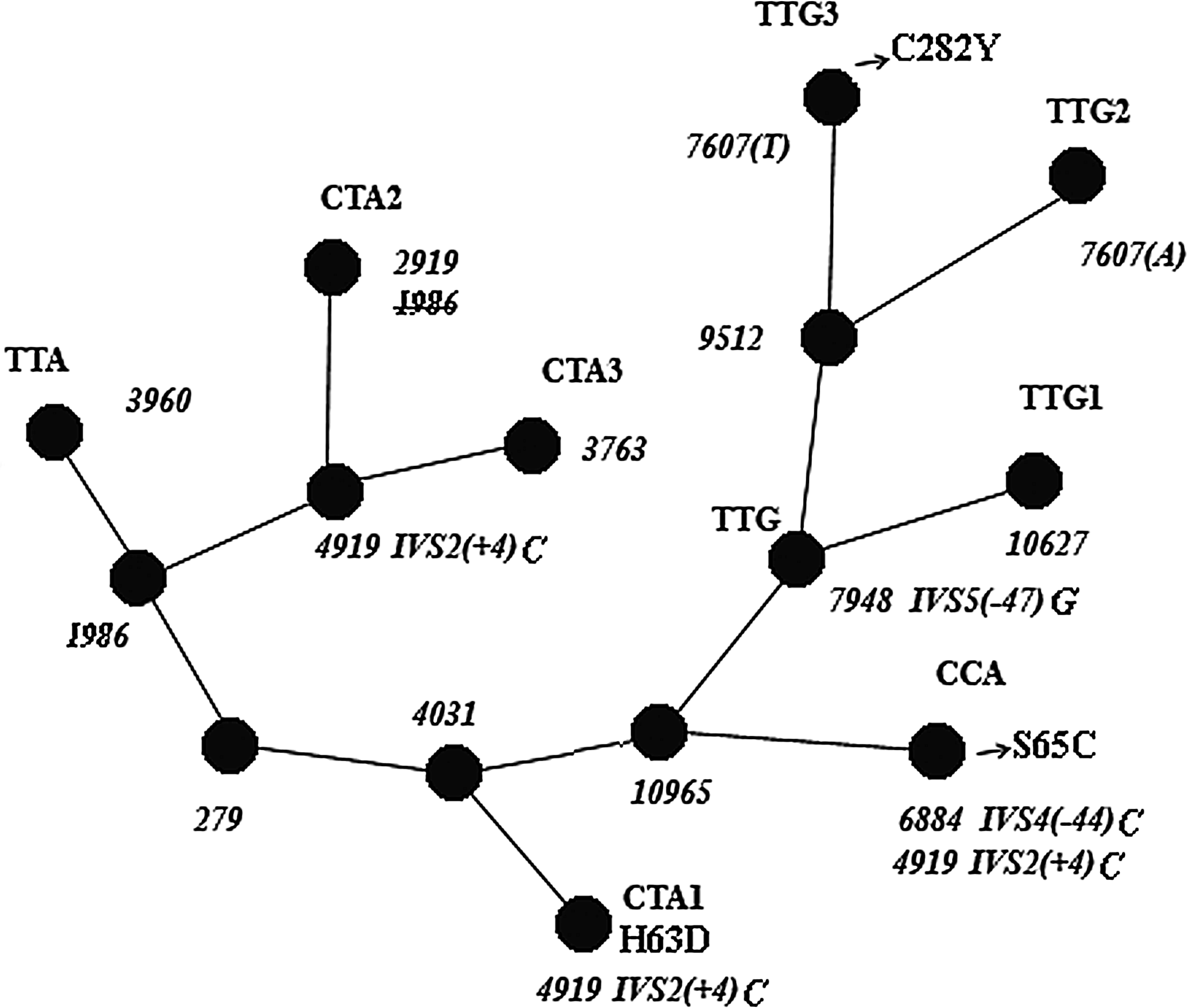
A minimal parsimony HFE tree showing relationships between the TTG, CTA, CCA, and TTA allelic sequences. Note: The common polymorphic sites for allele group sequences are numbered in the unrooted HFE gene tree as in Supplemental Figure S1.
The CTA1 sequences shared no substitutions with the CTA2 and CTA3 alleles, except the one at 4919 [IVS2(+4)] (Fig. 1, Supplemental Fig. S1). This is the most likely evidence of the independent origin of IVS2(+4)c in these two instances. All the TTG allele groups together with CTA1 and CCA sequences have common substitution at position 4031, and these share a substitution at 10965 with CCA allele only. This may be taken to mean that the CCA allele might have had an ancestral sequence in common with the TTG, but not with the CTA alleles. If so, substitutions at position 4919 [IVS2(+4)] in the CCA and CTA1 allele variants may be regarded as two events independent of each other. Thus, comparative HFE sequences analysis provided support for the assumed multiple independent IVS2(+4)t → c substitutions in the gene.
Clearly, the sequences with the different intronic haplotypes have considerably diverged from each other. Nevertheless, the alleles with the C282Y, H63D, and S65C mutations, associated with different intronic haplotype variants, contain the same region with identical substitutions at 782, 1436, and 2470 (Supplemental Fig. S1), comprising a promoter region of about 1 kb, exon 1, and a third or so of intron 1. This was explained by several independent conversion events in this DNA fragment. The allele variants from the TTG and CTA groups (Fig. 1) may obviously occur in different proportions in the human populations of Eurasia (Table 3).
DNA context analysis of the HFE intron 2, 4, and 5 regions with the IVS2(+4)t/c, IVS4(−44)t/c, and IVS5(−47)g/a sites: possible functions of these SNPs in splicing
Multiple factors had contributed to the current distribution patterns of different HFE alleles in the populations of Russia. Along with the founder effect and admixtures from subsequent migration flows, selective constraint might have affected the ethnospecific distribution of the HFE allele variants, if the SNPs in these noncoding regions are of functional importance. The location of the three polymorphisms in introns at a critical distance (shorter than 50 bp) from the splice sites increases the plausibility of this assumption (Sorek and Ast, 2003; Zhang et al., 2005).
IVS2(+4)t/c
The polymorphic site is located in the intron near the donor site junction and it is supposed to be able to affect the choice of the 5′ splice site (5′ss) during spliceosome assembly (Rogozin and Milanesi, 1997; Nagai et al., 2001; Lund and Kjems, 2002; Carmel et al., 2004). The human 5′ss sequences vary, but most correspond to the consensus A−2G−1/G1U2R3A4G5U6 (the R-purine nucleotide) (Lund and Kjems, 2002). The TTA and TTG allele variants contain t nucleotide at position + 4 in the HFE gene, the CTA and CCA allele variants have the rarer c nucleotide at this position in intron 2.
The 5′ terminal intronic heptamers function during the selection of concrete 5′ss in the target pre-mRNAs by U1 snRNP and then by U6 snRNP to form the active center of the spliceosome (Nagai et al., 2001; Lund and Kjems, 2002) (Supplemental Fig. S2, available online at www.liebertonline.com).
The correctness of a splicing procedure depends on base pair (bp) numbers in the 5′ss:U1 snRNA and the 5′ss:U6 snRNA complexes, because U6 snRNA removes U1 snRNA during spliceosome active center formation (Nagai et al., 2001; Lund and Kjems, 2002).
In the case of U+4, 5′ss could form 8 bp with U1 snRNA and 5 bp with U6 snRNA, and in the case of C+4, it could be 8 and 4 bp, respectively. Thus, removal of the U1 snRNA from the spliceosome could be complicated in the case of IVS2(+4)c and it may promote exon skipping.
The presence of C+4 in intron 2 in the CTA and CCA allele variants is hardly a chance event, because C is also present at this position in most of the HFE gene orthologs (Supplemental Table S3).
Among the HFE mRNA isoforms retrieved from GenBank, besides the full-length variants (accession no. U60319), there are some with skipping exon 2 (accession nos. AJ249336, AF079408, AF079409) and also variants lacking other exons. This is an indirect evidence that there may possibly exist different mechanisms for selection of the exon 2/intron 2 splice site. However, the role of these isoforms is presently unknown.
IVS4(−44)t/c
The HFE mRNA intron 4 is the classical “short” intron, is 158 bp long, and contains nucleotide purine blocks in higher abundance. Such blocks frequently occur in the binding sites for the various protein splicing factors (McCullough and Berget, 1997; Sorek and Ast, 2003; Yeo et al., 2007). Thus, there is reason for suggesting that intron 4 possesses specific regulatory features.
Among the HFE mRNAs with modifications of exon 4, there are variants differing by the location of the donor splice site (GenBank accession no. AF150664) or by an additional exon in the immediate vicinity to it (GenBank accession no. BC074721). The 5′ss sequences involved in the processing of these HFE pre-mRNAs have nucleotides (-gtaa-) at positions (+1) to (+4) corresponding to the consensus, and purine stretches in the exon portion of the donor splice sites (Table 4).
The single-nucleotide polymorphisms localized in the proximity to exon 4 and within intron 4 are shown in boldface. Position −1 of the sites are given according to GenBank sequence data (accession no. Z92910).
The 5′ splice site at the additional short 33-bp exon localized upstream from exon 4.
Intronic haplotypes marked according to Supplemental figure S1 are bracketed.
Context analysis of the intron 4 DNA fragment with the (−44) t/c polymorphism we studied suggests the possible presence of the putative 5′ss with this particular SNP at position −5 (potential 1, Table 4). Another 5′ss motif (potential 2, Table 4) is possibly present in this intron at a distance of 74 bp upstream from the potential 1 site. These data suggest that HFE pre-mRNA splicing may occur with the retention of intron 4 DNA fragments (Galante et al., 2004).
The structural conformity between the HFE mRNA isoforms is presented in Figure 2.
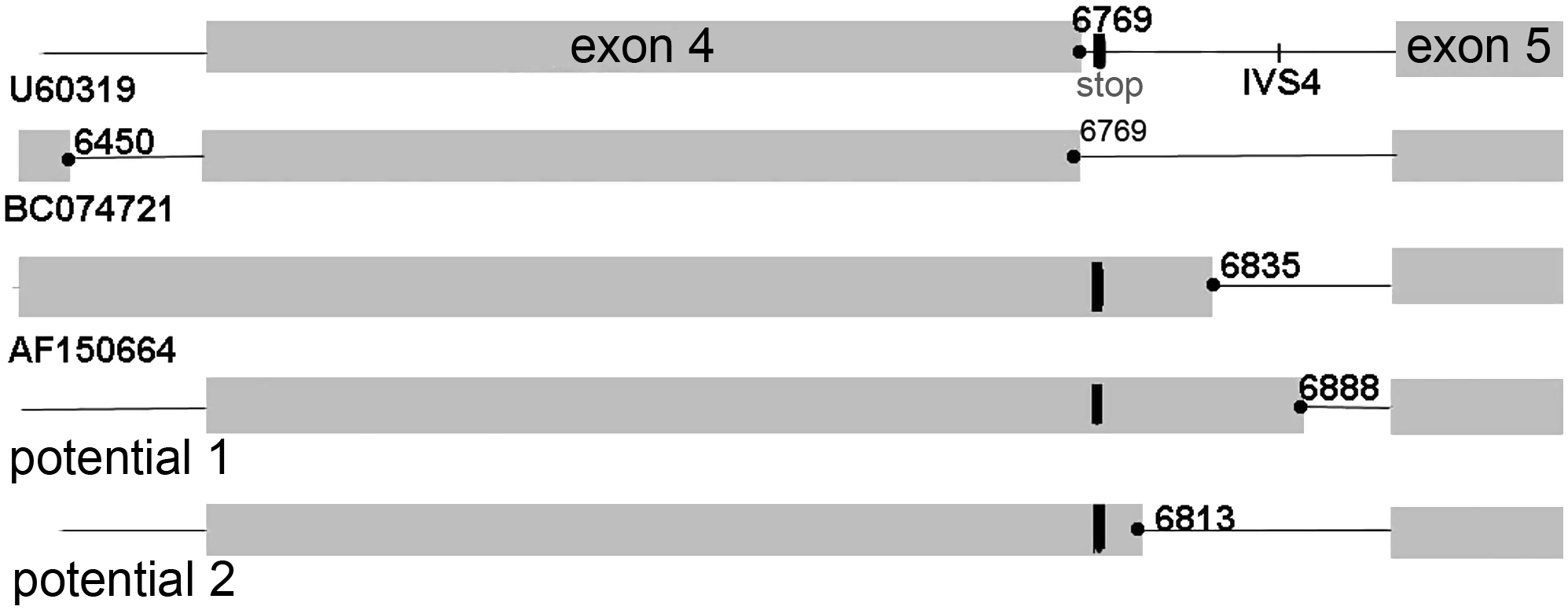
Schematic distribution of the (−1) positions in the observed and putative donor splice sites in a proximity of exon 4, according to Table 4. Note: Exons are shown in dark. STOP codon is located in frame with exon 4.
The mRNAs with intron 4 inserts could ultimately generate truncated soluble forms of the HFE protein by employing the stop codon at a distance of five nucleotides away from the exon 4/intron 4 boundary with conservation of the open reading frame. Expression of HFE mRNA with retained intron 4 and shortened protein has been observed in liver tissue (Jeffrey et al., 1999). Such a generation mode of a soluble protein has been demonstrated for the major histocompatibility complex, class I, G protein (HLA-G) as well (Fujii et al., 1994).
IVS5(−47)g/a
The polymorphic site is located within the nucleotide block -gcaagatg(
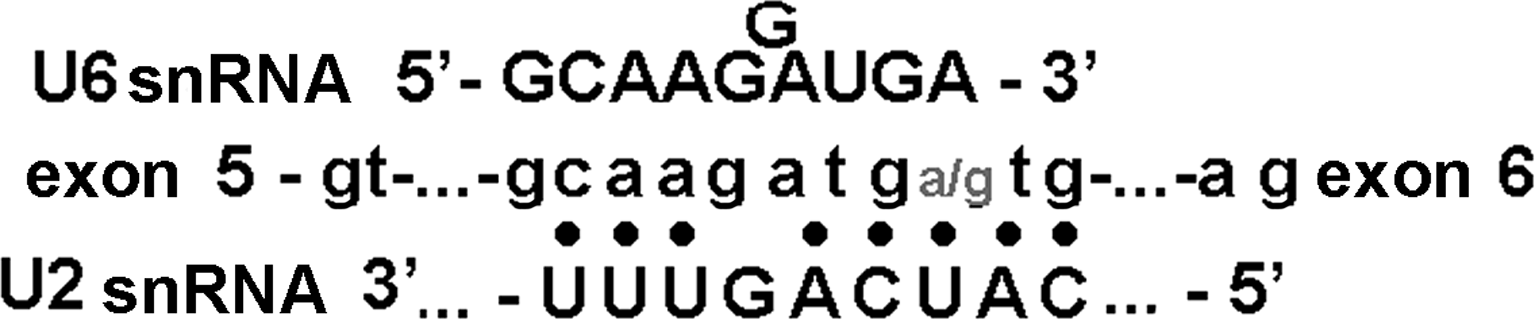
RNA:RNA interactions during spliceosome formation at the intron 5/exon 6 splice site in the HFE pre-mRNA.
Such a DNA sequence in the proximity to the 3′ss may facilitate the extrusion of U4 snRNA from its complex with U6 snRNA and its substitution by U5 snRNA. In addition, it may help to draw together the U2 RNA to the HFE pre-mRNA and at the same time to the U6 RNA for providing the network interactions in the spliceosome assembly (Guthrie and Patterson, 1988; Nagai et al., 2001; Lund and Kjems 2002).
Ten variants of genotypes with the TTG, TTA, CTA, and CCA alleles of the HFE gene occur at different frequencies among populations of Russia. Carriers of different genotypes may potentially express distinctive sets of alternative HFE mRNAs on the norm. Altogether, the wide range of individual phenotypic features of iron metabolism turnover in health and disease (Ludwiczek et al., 2004) may to a large measure be defined by the intronic haplotype allelic architecture in the genotype. This assumption needs supportive experimental evidence.
Footnotes
Acknowledgments
This research was supported by the Biodiversity and Gene Pool Dynamics grant from the Russian Academy of Sciences Presidium. The authors are grateful to anonymous referees for critically reading the manuscript. They also express their gratitude to A. Fadeeva for translating this article from Russian into English.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.