
Abstract
Samples from 30 deaf probands exhibiting features suggestive of syndromic mitochondrial deafness or from families with maternal transmission of deafness were selected for investigation of mutations in the mitochondrial genes MT-RNR1 and MT-TS1. Patients with mutation m.1555A>G had been previously excluded from this sample. In the MT-RNR1 gene, five probands presented the m.827A>G sequence variant, of uncertain pathogenicity. This change was also detected in 66 subjects of an unaffected control sample of 306 Brazilian individuals from various ethnic backgrounds. Given its high frequency, we consider it unlikely to have a pathogenic role on hereditary deafness. As to the MT-TS1 gene, one proband presented the previously known pathogenic m.7472insC mutation and three probands presented a novel variant, m.7462C>T, which was absent from the same control sample of 306 individuals. Because of its absence in control samples and association with a family history of hearing impairment, we suggest it might be a novel pathogenic mutation.
Introduction
H
There are many reports on novel mutations in mitochondrial DNA that could be responsible for hearing loss. In some cases, lack of population information leads to controversy on the pathogenic significance of these mutations (Yao et al., 2006). One of the most controversial questions regards the MT-RNR1 m.827A>G mutation (Li et al., 2005; Xing et al., 2006a, 2006b; Chaig et al., 2008).
To investigate the contribution of MT-RNR1 and MT-TS1 mutations to hearing loss in a Brazilian sample, we searched for mutations in 30 unrelated probands. Our work contributes to the discussion on the pathogenic effect of mitochondrial mutations on hereditary deafness.
Patients and Methods
Subjects
We selected 30 affected individuals for molecular investigation of mitochondrial genes, from genealogies with familial hearing loss, compatible with maternal inheritance (22 cases), or presenting symptoms frequent in syndromic forms of mitochondrial deafness, such as neurological disorders and diabetes (6 cases) or fulfilling both criteria (2 cases). These individuals were ascertained in our genetic counseling unit, Centro de Estudos do Genoma Humano, Universidade de São Paulo, Brazil. In all probands, the most frequent mutations associated with hearing loss, m.1555A>G in the mitochondrial MT-RNR1 gene, c.35delG and c.167delT in the nuclear GJB2 gene, and del(GJB6-D13S1830) and del(GJB6-D13S1854) in the GJB6 gene, had been previously excluded as being the cause of the hearing impairment.
A control sample was also screened for specific variants. It consisted of 306 Brazilian unaffected individuals of various ethnic backgrounds: 133 subjects were predominantly of European ancestry, 165 were of predominantly African ancestry, and 8 were of Asian ancestry. Part of the control sample is described in Abreu-Silva et al. (2010). The Brazilian population is highly admixed and subjects, with either European or African ancestry, may present high frequencies of Native American lineages of mitochondrial DNA (Bortolini et al., 1997; Alves-Silva et al., 2000; Marrero et al., 2005).
Written informed consent was obtained from all normal controls, patients, and their relatives participating in the study.
Methods
Total DNA was extracted from peripheral blood using standard procedures. The two mitochondrial genes, MT-RNR1 and MT-TS1, were sequenced in all 30 probands. The MT-RNR1 gene was amplified by polymerase chain reaction (PCR) in two fragments using the following primers: 5′ ATG TAG CTT ACC TCC TCA AAG C 3′ and 5′ TTA AGC TGT GGC TCG TAG TG 3′, and 5′ TGC TTA GCC CTA AAC CTC AA 3′ and 5′ GGT TTA GCT CAG AGC GGT CA 3′. For the MT-TS1 gene, the primers used were 5′ GGA TGC CCC CCA CCC TAC C 3′ and 5′ CCT ACT TGC GCT GCA TGT GCC 3′. Each fragment was purified and submitted to sequence analysis in the MegaBACE 1000 DNA Analysis System (Amersham Biosciences).
To determine whether the m.7462C>T and m.7472insC mutations in the MT-TS1 gene are in a homoplasmic or heteroplasmic state, PCR-restriction fragment length polymorphism (RFLP) analysis was performed with enzymes TaqI for m.7462C>T and XcmI for m.7472insC. For the latter mutation, the detection of the C insertion was obtained using a modified forward primer (5′ GAA GGA ATC GAA CC
The probands whose sequence analysis showed m.827A>G and m.7462C>T mutations had their mitochondrial haplogroup determined by sequencing of the hypervariable region I with primers and protocols previously described by Alves-Silva et al. (2000). Over 95% of the mitochondrial DNA lineages can be assigned by this method (Marrero et al., 2007).
The control sample was screened by PCR-RFLP for the m.827A>G (MseI), m.7462C>T (TaqI), and m.13263A>G (HincII) substitutions. The latter substitution allows the identification of individuals with haplogroup C. Individuals from the control sample with m.827A>G also had their hypervariable region I sequenced for haplogroup identification.
Results and Discussion
The results are summarized in Table 1.
Mutational analysis of the MT-RNR1 gene
The known m.961delTinsC(n), m.1095T>C, and m.1494C>T mutations in the MT-RNR1 gene were not detected in our probands. However, five probands presented the m.827A>G mutation. Aminoglycoside exposure was not reported in these cases. This mutation was first reported in a sporadic case of deafness, in 2004 (Li et al., 2004). In 2005, Li et al. analyzed 128 Chinese pediatric patients with aminoglycoside-induced and nonsyndromic hearing loss; they found the m.827A>G mutation in five patients, and considered it as a putative pathogenic mutation. Xing et al. (2006a, 2006b) studied two nonrelated Chinese families with hearing loss in which the same mutation was present in all maternal relatives, but not all the carriers of the mutation were affected. This supposed incomplete penetrance led the authors to conclude that m.827A>G mutation alone is not sufficient to produce the clinical phenotype, which would require the involvement of modifying factors. In 2008, Chaig et al. detected the same mutation in an Argentinean family with hearing loss after aminoglycoside treatment. The contribution of the variant m.827A>G to hearing loss is, however, controversial: indeed, together with m.15535C>T, this variant defines the subhaplogroup B4bd of the haplogroup B mitochondrial lineage (Bandelt et al., 2003; Kong et al., 2003; Tanaka et al., 2004; Yao et al., 2006), and as noted by Bandelt et al. (2005), a variant that defines a whole major or minor subhaplogroup is very unlikely to be a primary disease mutation. In fact, these studies that associated m.827A>G mutation to hearing loss were performed in Chinese and Argentinean families, and the haplogroup B was found in both East Asia and South America (Torroni et al., 1993; Kong et al., 2003; Mishmar et al., 2003). Moreover, in the study of Li et al. (2005), this variant was also found in 2 of 144 controls, but this issue was not subjected to further discussion.
The five probands carrying the m.827A>G mutation detected in our study had their mitochondrial haplogroup determined as haplogroup B, which is in agreement with previous haplogroup studies (Bandelt et al., 2003; Kong et al., 2003; Tanaka et al., 2004). The Argentinean family studied by Chaig et al. (2008) also had haplogroup B. Further, in our control sample composed of 306 individuals with normal hearing, we detected 66 (21%) individuals carrying this sequence variant. Interestingly, other haplogroups, in addition to haplogroup B, such as the Native American A and C and African haplogroups L1 and L3, were found to be associated with m.827A>G mutation among these 66 individuals, indicating it is not exclusive to subhaplogroup B4bd. Given its high frequency in the hearing population, it seems unlikely that m.827A>G is the cause of hereditary hearing loss, even with a low penetrance.
Mutational analysis of the MT-TS1 gene
The previously reported MT-TS1 mutations, m.7445A>G, m.7510T>C, and m.7511T>C, were not detected in our sample. One of the probands carrying an m.827A>G mutation in the MT-RNR1 gene also presented the known pathogenic m.7472insC mutation in the MT-TS1 gene, apparently in homoplasmy after PCR-RFLP and sequencing. The pedigree shows a pattern of transmission of hearing loss compatible with mitochondrial inheritance (Fig. 1). At clinical examination, a proband (IV-10) presented ataxia and sensorineural hearing loss, noticed at the age of 6 years. Her maternal aunt (III-4) presents deafness but no ataxia, and heteroplasmy was not detected after PCR-RFLP and sequencing. Other affected family members had their hearing loss detected approximately at the age of 25-30 years, and ataxia was not found either. This mutation was first described in 1995 (Tiranti et al., 1995) in six patients whose deafness was accompanied by ataxia, dysarthria, and focal myoclonus. Subsequently, it was identified in two subjects presenting ataxia, myoclonic epilepsy, and mental retardation (Jaksch et al., 1998). Therefore, though the proband also has the m.827A>G variant, her hearing impairment and ataxia are most probably due solely to m.7472insC mutation.
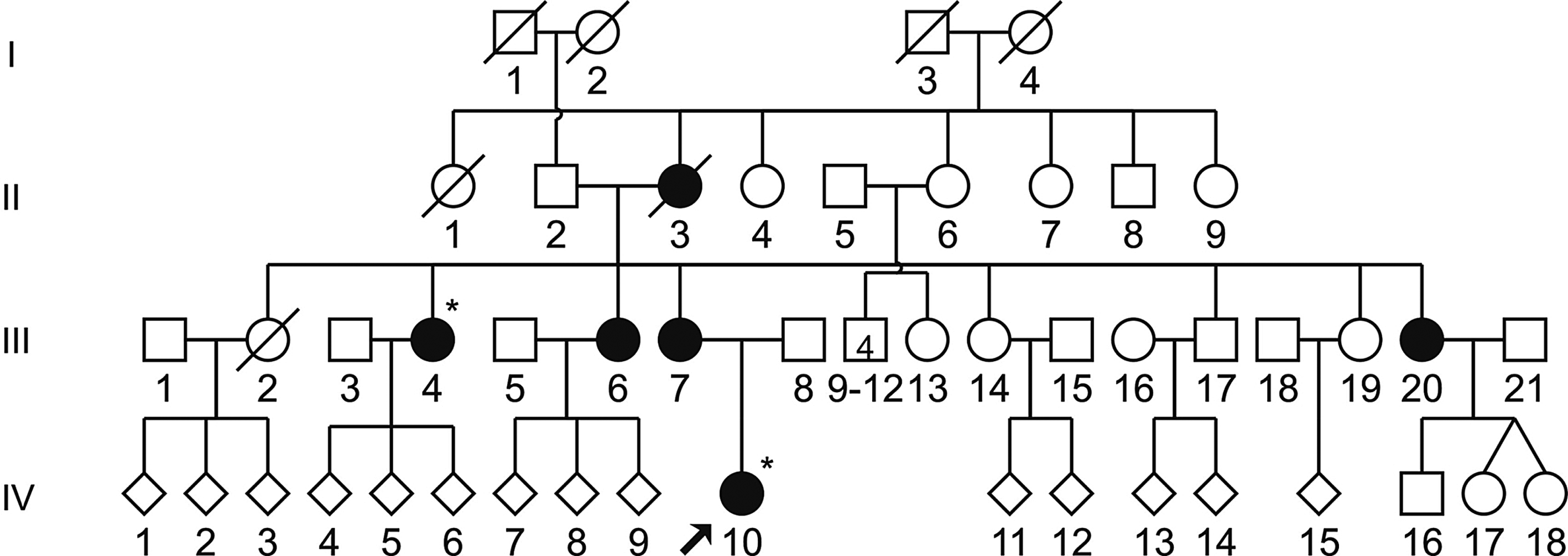
Pedigree of a family with m.7472insC mutation in the MT-TS1 gene. Hearing-impaired individuals are indicated by full symbols, arrow denotes the proband, and asterisks denote individuals with mitochondrial DNA studied.
We also detected a novel variant in the MT-TS1 gene, m.7462C>T (Fig. 2), in three unrelated probands; we did not detect heteroplasmy in either case after PCR-RFLP and sequencing. All probands had nonsyndromic hearing loss and belong to genealogies in which maternal inheritance of deafness was present (Fig. 3). Figure 4 shows tonal audiograms of all individuals with m.7462C>T who had their mitochondrial DNA examined. In one case (Fig. 3a), the proband (II-7) was examined at the age of 30 years, presenting bilateral, mild, and sensorineural hearing loss. Hearing loss was noticed at the age of 26 years. Her older daughter (III-4) presented bilateral, moderate-to-severe, and sensorineural hearing loss, whereas her younger daughter (III-5) presented bilateral, moderate, and sensorineural hearing loss (Fig. 4). In the second case (Fig. 3b), the proband (IV-3) was examined at the age of 14 years and also presented bilateral, moderate, and sensorineural hearing loss, whereas his mother (III-5) has mild-to-moderate hearing loss. Finally, the third proband carrying the m.7462C>T mutation (Fig. 3c; V-3) was examined at the age of 6 years and presented bilateral moderate hearing loss. Onset of deafness was postlingual in all cases.

Partial sequence analysis of MT-TS1 gene in the 7462 region. (
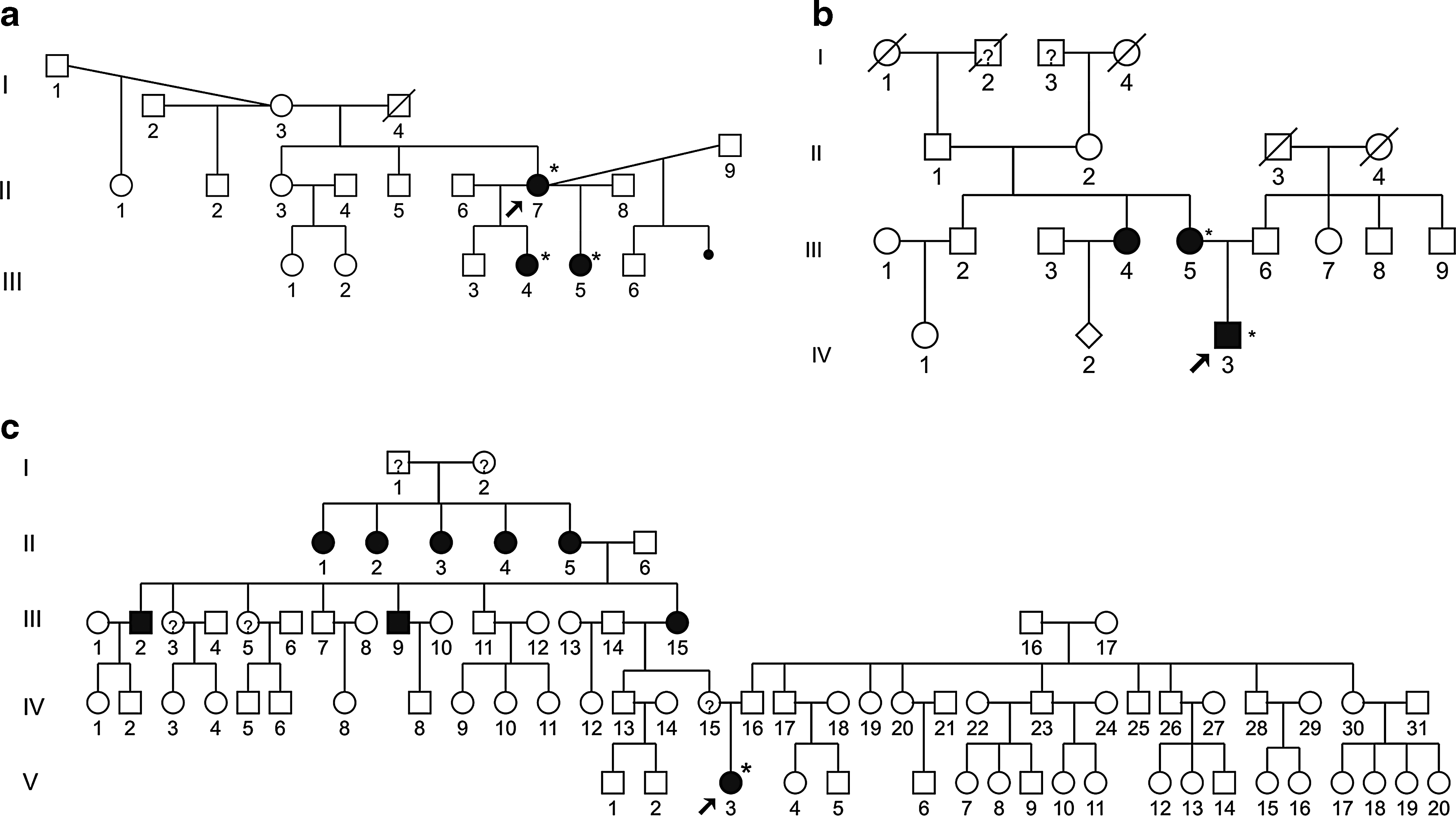
Pedigrees
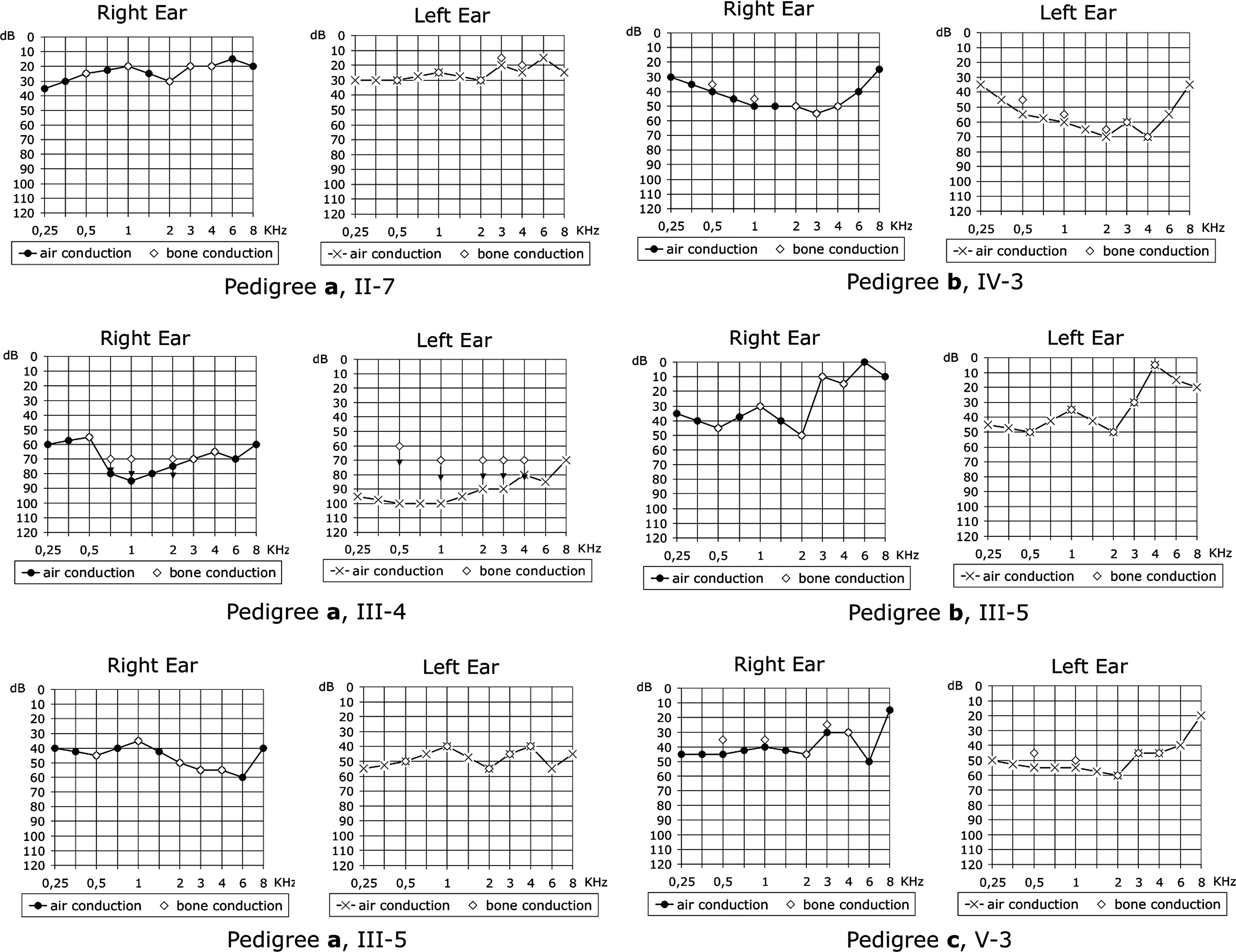
Tonal audiograms of the individuals with m.7462C>T mutation in the MT-TS1 gene, from pedigrees
The m.7462C>T mutation was not previously reported in databases such as Mitomap (2009), mtDB (2009), and GenBank (2009). We determined the mitochondrial haplogroups and found that all three probands had mitochondrial DNA of haplogroup C of Native American origin. Therefore, the m.7462C>T variant could be a polymorphism of haplogroup C. However, this variant has never been reported as a polymorphism in the few studies of the coding region of mitochondrial genes in haplogroup C (Tamm et al., 2007; Achilli et al., 2008). In addition, our control sample of 306 subjects (58 of which were classified in haplogroup C) was tested for m.7462C>T with restriction enzyme TaqI and none presented this mutation. These data strongly suggest that the m.7462C>T variant is rare and might be either a very rare polymorphism of haplogroup C or a novel pathogenic mutation leading to hearing impairment.
It is a hard task to speculate on mechanisms that lead to phenotype when the product is a transfer RNA that does not allow a precise prediction of the effect of a substitution. According to McFarland et al. (2004), two criteria have to be considered when assessing the pathological significance of a novel mitochondrial tRNA variant: the location of the mutation in the secondary structure of the tRNA molecule and the disruption it causes to Watson-Crick base pairing. By comparing the characteristics of neutral variants with those of pathogenic mutations, they concluded that the hotspots for pathogenic mutations occur in stems, especially in the acceptor and anticodon stems. The 7462 position in the mitochondrial tRNASer(UCN) molecule is located at the TΨC loop; however, it is also the case of the pathogenic mutations m.3291T>C in the tRNALeu(UUR) gene (MT-TL1) and m.8344A>G in the tRNALys gene (MT-TK). So, its location does not rule out the possibility of being pathogenic.
In summary, our data suggest that the m.827A>G variant in the MT-RNR1 gene is unlikely to be pathogenic. The m.7462C>T variant in the MT-TS1 gene might appear as a mutation leading to hearing impairment. Nevertheless, the definitive classification of m.7462C>T as a pathogenic variant requires further investigation, namely the finding of this variant in mitochondrial genomes belonging to haplogroups other than haplogroup C, as well as functional studies to assess its effects on the tRNASer(UCN) molecule. Our results also reflect the importance of using a suitable population approach in clinical studies, to avoid premature claims as to the pathogenic role of mutations. Such studies are particularly appropriate in an admixed population such as the Brazilian.
Footnotes
Acknowledgments
The cooperation of all the hearing-impaired patients, their relatives, and all the control individuals is acknowledged. The authors thank the professionals from DERDIC for helping in the clinical evaluation of individuals. The authors also thank Dr. Paulo A. Otto, Dr. Angela Vianna-Morgante, and Lilian Dluhosch for critical reading of the manuscript. This research was supported by CEPID-FAPESP and CNPq.
Disclosure Statement
No competing financial interests exist.