
Abstract
We describe epidemiological, genetic, and clinical data of the 1124-2del mutation in the G6PT gene, detected in homozygosity in three glycogen storage disease type Ib patients of Sardinian origin. This mutation was found to be associated with four sequence variations: c.593 A>T (p.N198I), c.625+19 C>T, c.1062 C>T (N354N), and c.1224 G>A (p.T408T) in the G6PT gene. RNA studies were performed for c.1124-2del and c.625+19 C>T. The c.1124-1del2 acceptor splicing mutation showed skipping of 31 nucleotides of exon 9 due to the activation of a downstream cryptic acceptor splice site in 1154-1155 nucleotide positions, resulting in a downstream stop codon at aa position 402. RNA analysis of c.625+19 C>T variation showed a small amount of alternative splicing with skipping of exon 4, resulting in a stop codon at aa position 211. Our cases present most of features of the severe form of disease, including early onset with chronic neutropenia, frequent infections, and inflammatory bowel disease. Our results suggest a founder effect for glycogen storage disease type Ib that facilitates diagnosis using mutation analysis, sparing patients from liver biopsy. DNA-based diagnosis will enable us to make accurate determination of carrier status and prenatal diagnosis, thus improving genetic counseling.
Introduction
G
In this study, we report clinical data and molecular characterization with DNA and RNA analyses of three GSD Ib patients of Sardinian origin, suggesting a founder effect for GSD Ib in this population.
Patients and Methods
Patients
Three Sardinian patients with apparently nonconsanguineous parents and affected by GSD Ib were studied. In two patients, diagnosis was performed based on clinical and biochemical analyses including liver biopsy. In one of them, molecular analysis was previously performed and mutation detection further confirmed the disease (Veiga-da-Cunha et al., 1998). In the third patient, the presence of GSD Ib was suspected on the basis of clinical examination and biochemical tests, and mutational analysis was taken under consideration to confirm diagnosis, to avoid liver biopsy.
DNA analysis
Genomic DNA was isolated from peripheral blood leukocytes using standard methods. Informed consent was obtained from the proband's parents and from normal controls. DNA-PCR analysis was performed on exons 4, 8, and 9 and intron/exon boundaries of the G6PT1 gene as previously described (Veiga-da-Cunha et al., 1998), using appropriate pairs of primers (primers available upon request). Single strand conformation polymorphism (SSCP) analysis of a small aliquot of the amplified products was carried out as previously described (Loudianos et al., 1999a). Direct sequencing of the shifted exons was performed using dGTP technology and the ABI 3730 analyzer (Applied Biosystems Perkin-Elmer Corporation) according to the manufacturer's recommendations. The sequencer software package (version 4.2; GeneCodes Corporation) was used for sequence analysis. DNA haplotype analysis was performed in control families of Sardinian origin (father, mother, and sib) for the detected sequence variations using the SSCP method. The effect of mutation N198I in the G6PT gene was assessed in combination with clinical phenotype and by in silico analysis using “PolyPhen” and “Pmut.”
RNA studies
RNA analysis for the c.1124-1del2 and c.625+19 C>T mutations was performed on total RNA extracted from peripheral lymphoblasts of one homozygote, one heterozygote, and a normal control. RNA extraction was performed using the Ultraspec™ A reagent (Biotecx) according to the manufacturer's recommendations. RT-PCR was carried out with the GeneAmpRNA PCR Core kit (Perkin Elmer) according to the manufacturer's instructions.
First-round RT-PCR was performed in 100 μL volumes on 300 ng of total RNA. A second round of PCR was carried out using 1 μL of the first PCR product as template and appropriate nested primers. The primers used (sequences available upon request) were P1 and P2 located in exon 7 and the 3′ UTR, respectively, for the first-round reactions and P3 located in exon 8 associated to P2 in the nested PCR for c.1124-1del2 analysis (Fig. 1C); P4 and P5 located in exons 3 and 7, respectively, for the first-round PCR and P4 and P6 on exons 3 and 6, respectively, in the nested PCR for the c.625+19 C>T substitution (Fig. 1E). The amplified products were separated on 2% agarose gel and the fragments were cut and purified using QIA Quick columns (Qiagen) gel extraction kits, according to the manufacturer's protocol. Sequencing was performed as described earlier.
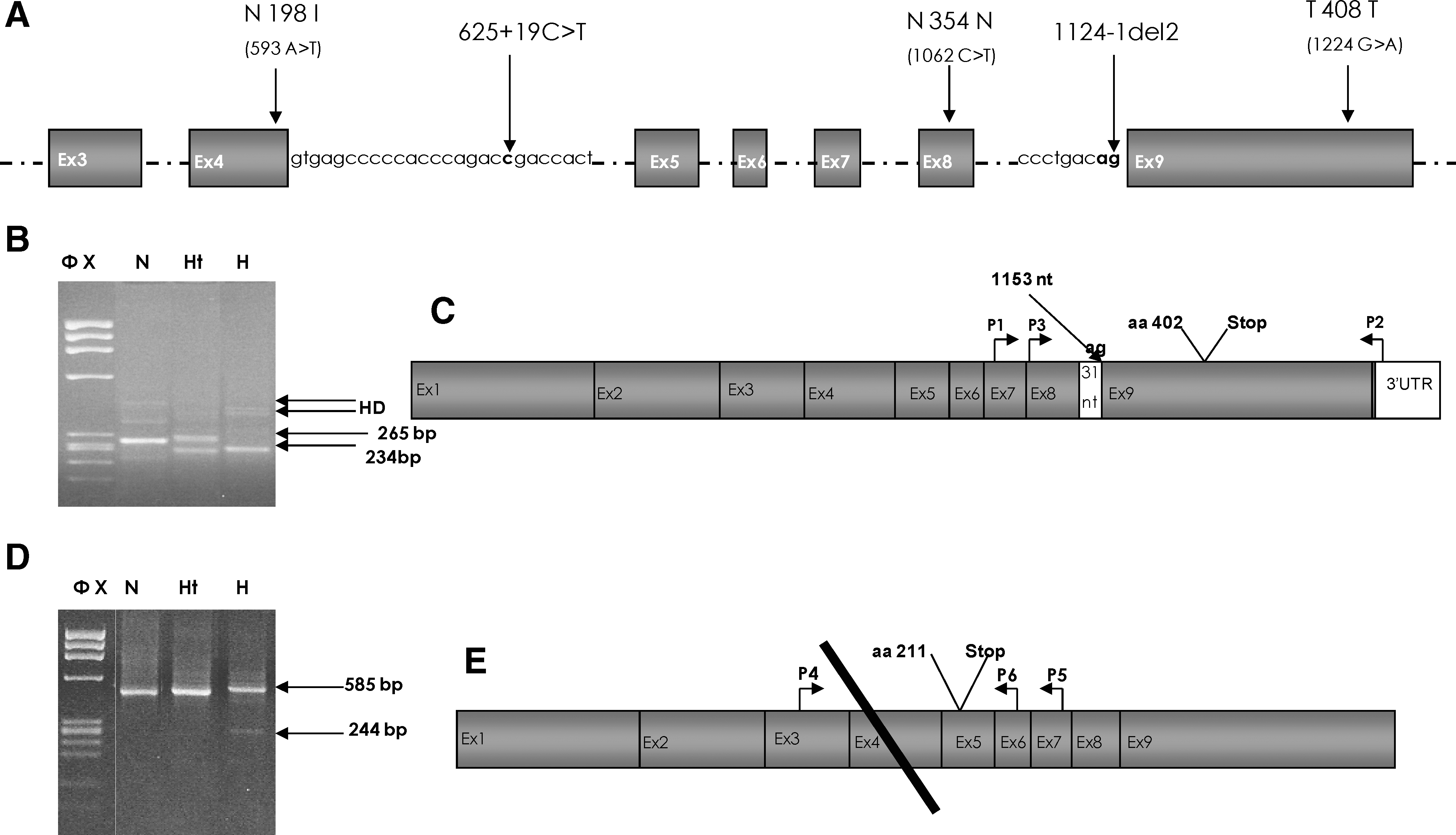
Schematic representation of the SLC37A4 gene and RT-PCR analysis of the 1124-1del2 and 625+19 C>T mutations. (
Results
DNA analysis
Mutation analysis was performed on exons 4, 8, and 9 of G6PT1 gene where, according to a previous paper, the 1124-1del2 and the three sequence variations c.593 A>T (p.N198I), c.1224 G>A (T408T), and c.1062 C>T (N354N) were detected in a patient of Sardinian origin affected by GSD Ib (Veiga-da-Cunha et al., 1998). Our results confirmed the presence of all the previously described sequence variations in the homozygous state in all three analyzed patients (Fig. 1A). In addition, a new sequence variation in intron 4, c.625+19 C>T, was also detected in homozygosity in all three patients (Fig. 1A). All identified sequence variations segregate in linkage and, with the exception of c.1224 G>A (T408T) (Pan et al., 1999), are population specific because they have been described in only the Sardinian GSD Ib patients.
The screening of 180 chromosomes of Sardinian origin showed that c.625+19 C>T and c.1224 G>A are relatively frequent. The c.625+19 C>T was detected in 17 of 180 (9.4%) chromosomes, in three cases in homozygosity, and the c.1224 G>A (T408T) was detected in 28 of 180 (15.5%). The c.1062 C>T (N354N) was found to be rare, being detected in 8 of 180 (4.4%), whereas c.593 A>T (p.N198I) was detected in only 5 of 180 (2.7%) chromosomes. We constructed haplotypes into which these sequence variations assemble, finding that c.593 A>T (p.N198I) occurs on haplotype IX present in one chromosome and on haplotype V (Table 1) present in three chromosomes and is associated with c.1124-1del2 (IVS8-1del2) on haplotype VIII in an additional chromosome. These data suggest that c.593 A>T (p.N198I) belongs to a rare haplotype where at a second time the c.1124-1del2 occurred. As c.593 A>T (p.N198I) was detected in the heterozygous state in all four samples, it was difficult to clarify its contribution in the disease status. This missense substitution replaces a neutral polar residue with an hydrophobic one. Using Blast search (www.ncbi.nih.gov/BLAST), we observed that asparagine is not conserved in homologous proteins in other transporters of phosphorylated metabolites. Both outputs from the in silico analysis predicted the mutation to be pathologic (PolyPhen: probably damaging with a score of 2.298; Pmut: pathological with a reliability index of 1).
RNA analysis
To better understand the mechanisms and role played by c.625+19 C>T and the acceptor c.1124-1del2 mutation in the disease phenotype, we performed RNA studies using total RNA from peripheral lymphoblasts. Agarose gel electrophoresis of the nested PCR products using the sense primer localized in exon 8 and the antisense primer localized in the 3′ UTR region in the nested PCR for c.1124-1del2 mutation showed a 265 bp normal band in the heterozygote and normal control sample and a 234 bp abnormal minor product in the homozygote and heterozygote samples (Fig. 1B, C). Sequence analysis of the minor product revealed a 31 bp deletion spanning from nucleotides 1124 to 1155 of exon 9. This alternative splicing product resulted in partial skipping of exon 9, creating a frameshift and a downstream stop codon at aa position 402 (Fig. 1C). The predicted truncated short protein is most likely not functional because it lacks the Tm9 region, including the LL dinucleotide, which is considered the signal for protein retention. The mechanism producing this alternative splicing is due to the deletion of the canonical acceptor splice site of intron 8, which activates a cryptic splice site AG in nucleotide positions 1154-1155. Agarose gel electrophoresis using the sense primer P4 and the antisense P6 localized in exons 3 and 6, respectively, in the nested PCR for the c.625+19 C>T substitution showed a 585 bp normal major product in all homozygote, heterozygote, and normal samples and a 244 bp abnormal minor product in only the homozygote samples (Fig. 1D, E). Sequencing analysis of the 244 bp band showed an exon 4 skipping that creates a frameshift and a stop codon at aa position 211, most likely resulting in a short, truncated, nonfunctional protein lacking the Tm3-Tm10 domains (Fig. 1E).
Discussion
In this study, we report clinical data and molecular characterization with DNA and RNA analyses of three Sardinian patients affected by GSD Ib. One of them was previously analyzed and found to be homozygous for the acceptor site splicing deletion mutation c.1124-1del2 in intron 8 (IVS8-1del2) in the homozygous state (Veiga-da-Cunha et al., 1998). In this study, we confirmed this result and detected the same mutation in two more patients. We also confirmed the association of c.1124-1del2 with four sequence variations, c.593 A>T (p.N198I), c.625+19 C>T, c.1062 C>T (N354N), and c.1224 G>A (T408T), in all the patients studied.
RNA studies suggested that GSD Ib was produced in these patients by an alternative splicing mechanism. It is due to the presence of c.1124-1del2 mutation, which results in the production of a truncated protein lacking the Tm9 region, including the LL amino acids, which are considered the signal for protein retention (Fig. 1B, C). The resulting alternative isoform is predicted to be nonfunctional because it lacks a domain that is critical for normal protein localization and function. c.1124-1del2 is a determinant of disease status, whereas c.625+19 C>T is expected to have only a minor insignificant contribution, because it results in the production of a very small amount of a truncated 211-aa protein (Fig. 1D, E). This hypothesis is supported by the clinical information obtained from 2 of 3 homozygous subjects for c.625+19 C>T sequence variation as they appeared normal in a diagnostic approach for GSD Ib involving family history, physical examination, and specific laboratory tests (data not shown). Although we have some functional information for the splicing variants, no information is available for the c.593 A>T (p.N198I) missense substitution. According to the Blast search, it seems not to be pathogenic. However, in silico analysis predicted the mutation to be pathologic. Based on these data, further studies with functional assays are necessary to better clarify the nature of the N198I substitution.
Mutation studies of the G6PT1 gene have shown that there is an allelic heterogeneity for GSD Ib, with the presence of a small number of relatively frequent and a large number of rare mutations. There is some ethnic variability. In Caucasian patients, c.1042delCT (31%) and p.G339C (15%) are the prevalent mutations, accounting for over 40% of all cases, whereas in Japanese patients, p.W118R is the prevalent mutation, accounting for 44% of the alleles (Chou et al., 2002; Kojima et al., 2004).
Our results suggest that in Sardinia, GSD Ib is caused by only one mutational event, further suggesting that Sardinia is a founder population as has been already suggested by molecular analysis of other genetic diseases such as β-thalassemia, APECED, and Wilson's disease (Rosatelli et al., 1992, 1998; Loudianos et al., 1999b). This high allelic homogeneity existing in our population allows us to carry out some genotype-phenotype correlation considerations. Our cases present most of the features of the severe form of disease. All patients showed early onset with febrile metabolic acidosis, hypoglycemia, and neutropenia (Table 2). Two out of three patients showed short stature; mental retardation was not observed, but hyperlipidemia was present in all of them. Further, two older patients (PI and CM) showed obesity, Crohn's-like disease, and proteinuria, which were not present in the 3-year-old patient (FV), probably because of age. Only one (CM) showed liver adenoma.
G-CSF, granulocyte colony-stimulating factor.
In conclusion, the data obtained in this study reveal the presence of only one molecular defect in patients with GSD Ib, thus facilitating the diagnosis using c.1124-1del2 mutation analysis and sparing patients from liver biopsy. DNA-based diagnosis will enable us to make accurate determination of carrier status and prenatal diagnosis, thus improving genetic counseling. Further, the c.625+19 C>T sequence variation could potentially act by modifying the phenotype in GSD Ib in patients with mild mutations, or by contributing to the disease status in complex diseases such as nonalcoholic liver disease, neutropenia, and Crohn's disease. More studies with a large number of patients and controls will likely be necessary to demonstrate this hypothesis.
Footnotes
Disclosure Statement
The authors disclose no conflicts.