
Abstract
Introduction: We detected false homozygosity at the NESP55 differentially methylated region of the imprinted GNAS locus while analyzing the segregation of single-nucleotide polymorphisms (SNPs) in families with pseudohypoparathyroidism type Ib (PHP-Ib). We hypothesized that differential methylation of NESP55 could affect polymerase chain reaction (PCR) amplification, resulting in allele dropout. Methods: We genotyped 10 normal controls for four SNPs in NESP55 differentially methylated region. SNPs were amplified by standard PCR conditions and with the addition of dimethyl sulfoxide. The methylated allele was identified by HpaII analysis, and haplotypes were confirmed using subcloning strategies. All SNPs were also genotyped in a PHP-Ib patient (P1), carrying methylation at both NESP55 alleles, and in an in vitro methylated control DNA (SSSI-N4). Results: In the control samples, we identified allele dropout of the methylated allele in 85% of the amplifications, using standard PCR conditions. Addition of dimethyl sulfoxide to the PCR successfully prevented dropout in all cases. No amplification bias was observed for P1 and SSSI-N4 samples. Conclusions: For the first time, we report that differential methylation of imprinted regions can lead to preferential amplification of unmethylated alleles. Addition of coadjuvants to the PCR may facilitate amplification of both alleles, providing an accurate genotyping in cases with methylation-related diseases.
Introduction
G
Genetic defects, such as uniparental disomies, large deletions, duplications, or mutations, encompassing the imprinted genes or their control regions are usually responsible for imprinting syndromes (Amor and Halliday, 2008). Mapping and identifying such alterations in patients often requires the genotyping of microsatellite markers or single-nucleotide polymorphisms (SNPs) located across these regions. Polymerase chain reaction (PCR) is a widely used technique for the amplification of DNA sequences, before genotyping and/or mutation screening, through automated sequencing and/or restriction enzymatic analyses. The preferential amplification of one allele (allele dropout) in a heterozygous sample may result in incorrect genotyping, such as false homozygosity, or misdetection of mutations. Allele dropout may arise from differences in the GC content between alleles (that lead to differential denaturation), differences in allele size, mismatches between primer and template DNA resulting from SNPs in the primer-binding site (Walsh et al., 1992), or even low amounts of DNA (Piyamongkol et al., 2003).
We detected allele dropout at NESP55 DMR of the GNAS locus while analyzing the segregation of SNPs in families with PHP-Ib and we hypothesized that this artifact could be the result of the distinct allele methylation status. Therefore, in this article, we sought to evaluate the effect of differential methylation of the NESP55 DMR alleles on the efficiency of PCR amplification. We genotyped SNPs located in this DMR (Fig. 1) in the DNA from 10 control individuals and detected false homozygosity due to low/absent amplification of the methylated allele. Addition of high concentrations of dimethyl sulfoxide (DMSO) to the PCR successfully prevented allele dropout in all cases.
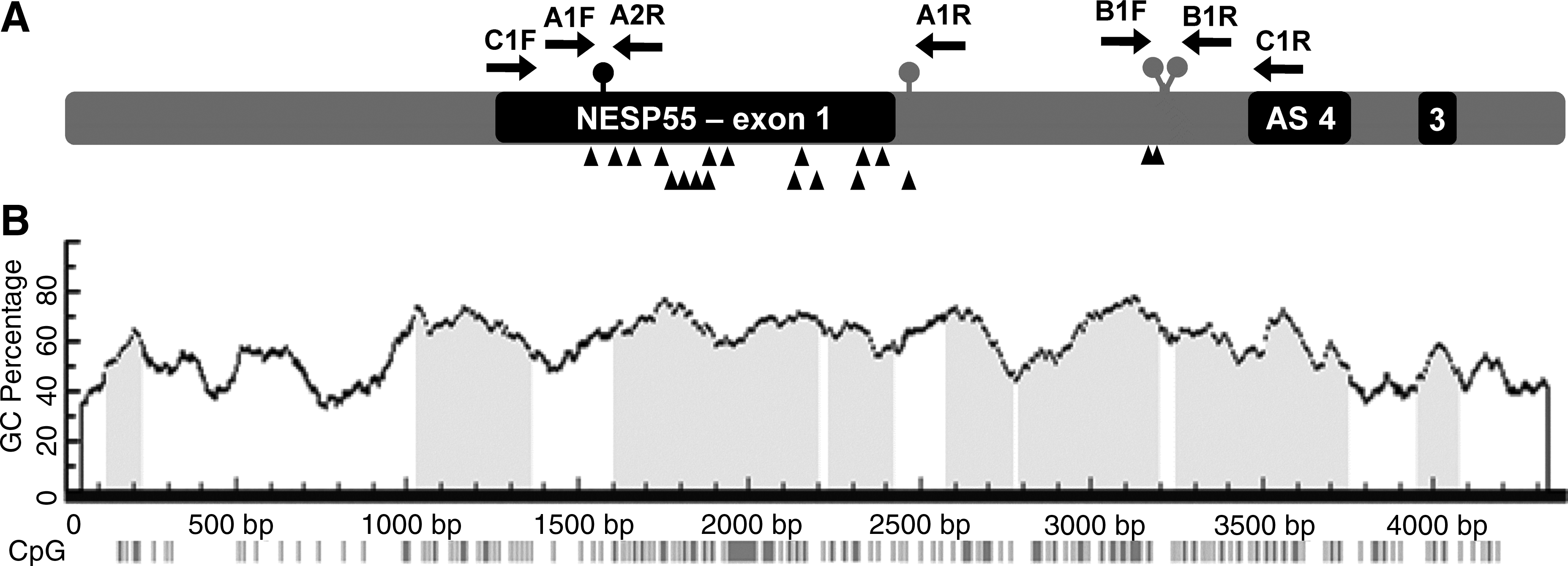
Schematic representation and cytosine-phosphate-guanine (CpG) island prediction in NESP55 DMR. (
Materials and Methods
This study was approved by our institution ethical committee. Venous blood was obtained following patient's written informed consent. Genomic DNA from a patient with PHP-Ib (P1) and from 15 normal controls was extracted from peripheral blood leukocytes, using standard protocols. For P1 and controls, two DNA fragments were amplified in the NESP55 DMR: segment A (encompassing rs1800900 and rs1800905) and segment B (including rs55995056 and rs3787497). Standard conditions for the amplification of these fragments were 50 ng of DNA, 1× PCR buffer, 1.8 mM MgCl2, 200 μM dNTPs, 0.4 μM of each primer, and 0.5 U Taq DNA polymerase (Invitrogen, Paisley, PA, United Kingdom) in a reaction volume of 12.5 μL. Amplification of segments A and B was also performed with the addition of DMSO (Merck, Darmstadt, Germany) to the reaction at 3% and 6% (v/v). PCR conditions were 95°C for 5 min, followed by 35 cycles, consisting of denaturation at 95°C for 45 s, annealing at 59°C for 35 s, and extension at 72°C for 40 s, followed by a final elongation step at 72°C for 10 min. Primer details and product GC content and size are indicated on Table 1.
Primers were designed with Primer3 (Rozen and Skaletsky, 2000).
Nucleotides from GenBank sequence AL132655.
Amplification primers also used for sequencing.
Primer used only for sequencing.
SNP, single-nucleotide polymorphism; Tm, melting temperature.
Sequencing reactions were performed using the ABI Prism Big Dye Terminator Cycle Sequencing Ready Reaction Kits (Applied Biosystems, Foster City, CA) with Ampli Taq DNA polymerase. Reactions were run on an ABI Prism 310 Genetic Analyzer (Applied Biosystems) and analyzed with the Sequencing Analysis 3.4.1 software (Applied Biosystems). Sequence variations in the aforementioned SNPs were analyzed with VarDetect software (Ngamphiw et al., 2008) using the default settings (threshold heterozygosity level = 0.30; primary peak = 1.0) and other heterozygosity levels (0.50, 0.75, and 0.90).
DNA from the 10 control samples that were most informative and from P1 was digested with the methyl-sensitive enzyme HpaII (New England Biolabs, Ipswich, MA) at 37°C for 20 h. The efficiency of the digestion was monitored by gel electrophoresis. In vitro methylation of DNA from one normal control was performed with the site-specific CpG methyltransferase SSSI (New England Biolabs) at 37°C for 4 h. The efficiency of the reaction was monitored by HpaII digestion, followed by gel electrophoresis.
To confirm the haplotypes for the 10 normal controls, a 2.1-kb fragment encompassing the four SNPs (segment C) in the NESP55 DMR was amplified, with a different set of primers, and subcloned, allowing the sequencing of the maternal and paternal alleles separately. Segment C was amplified, in each DNA, with the LongRange PCR Kit (Qiagen, Hilden, Germany) using the following conditions: 50 ng of DNA, 1× PCR buffer, 3.5 mM MgCl2, 1× Q-Solution, 500 μM dNTPs, 0.4 μM of each primer, and 0.5 U of Enzyme Mix, in a reaction volume of 12.5 μL. Cycling conditions were 93°C for 3 min, followed by 35 cycles consisting of denaturation at 93°C for 15 s, annealing at 59°C for 30 s, and extension at 68°C for 4 min, and a final elongation step at 68°C for 10 min. The amplified products were inserted into the vector pCR 2.1-TOPO (Invitrogen) and then propagated in TOP 10 competent cells (Invitrogen). Colonies were grown under ampicillin selection and plasmid DNA was isolated. The four SNPs were sequenced in each plasmid DNA, as mentioned earlier, and the haplotypes in each chromosome of the normal controls were identified.
Results
The results of sequencing analysis for 10 control samples (N1-N10), which were more frequently heterozygous for the four SNPs, are summarized in Table 2. The remaining five control samples, which were less informative for these SNPs, were excluded from further studies. Using standard conditions (no DMSO) in NESP55 fragments A and B amplification, none of 10 samples were scored as heterozygous for rs1800900 and rs1800905, 2 of 10 samples were scored as heterozygous for rs55995056, and 3 of 10 for rs3787497. With addition of DMSO to the PCR, at a final concentration of 3% (v/v) (3% DMSO), 8 of 10 samples showed heterozygosity for rs1800900, 7 of 10 for rs1800905, 4 of 10 for rs55995056, and 7 of 10 for rs3787497. Addition of 6% DMSO resulted in the detection of all 10 heterozygotes for rs1800900, rs1800905, and rs3787497, and 5 of 10 for rs55995056. For the 10 control samples, genotypes for one marker (rs1800905; alleles A/G) were further confirmed through enzymatic hydrolysis with BstUI (CG^C
SNPs scored as heterozygous are indicated with the nucleotide of the primary peak first, followed by the nucleotide of the secondary peak, together with the maximum level at which heterozygosity was detectable in VarDetect (0.30, 0.50, 0.75, and 0.90).
Homozygotes with a visible secondary peak that was less than 30% of the primary peak.
Consensus genotypes were obtained after sequencing of the subcloned segment C.
SNP, single-nucleotide polymorphism.
To identify the methylation status of the dropped allele, segments A and B were also amplified with 6% DMSO, using HpaII-digested DNA. With this method, only the methylated alleles (uncleaved by HpaII) were expected to be amplified by PCR. The allele that either failed to amplify or yielded the secondary peak, following standard amplification from genomic DNA, was shown to be the methylated one in all 10 control samples for rs1800900, rs1800905, and rs3787497 and in 5 of 10 samples for rs55995056 (Table 2). This suggested that the control samples N2, N3, N6, N7, and N10 were true homozygotes for rs55995056. Thus, a 6% DMSO concentration resulted in the detection of 100% of the heterozygotes for all NESP55 DMR SNPs, whereas 85% (17/20) of amplifications with standard PCR conditions resulted in allele dropout.
To confirm the haplotypes for each normal control, a fragment encompassing NESP55 DMR (segment C) was amplified, using a kit containing a coadjuvant solution. These products were then subcloned, so that the maternal and paternal alleles could be individually sequenced, allowing the identification of each inherited haplotype and the consensus genotypes, which are represented in Table 2.
Amplification of segments A and B was also tested in patient P1 with PHP-Ib, who carried methylation at both NESP55 DMR alleles. Amplification from HpaII-digested DNA, with 6% DMSO, confirmed methylation at both alleles, because heterozygosity was detected at rs1800900, rs1800905, and rs3787497. Interestingly, amplification of segments A and B from P1 genomic DNA was also successful without DMSO addition, resulting in the detection of heterozygosity and thus suggesting that, when the two alleles are methylated, the amplification efficiency is equivalent for both (Fig. 2). To reproduce these results in vitro, fragments A and B were amplified from an SSSI-treated control DNA sample (N4), using standard PCR conditions. Both products were successfully amplified, and heterozygosity was detected in all SNPs (heterozygosity levels >0.50) (Fig. 2).
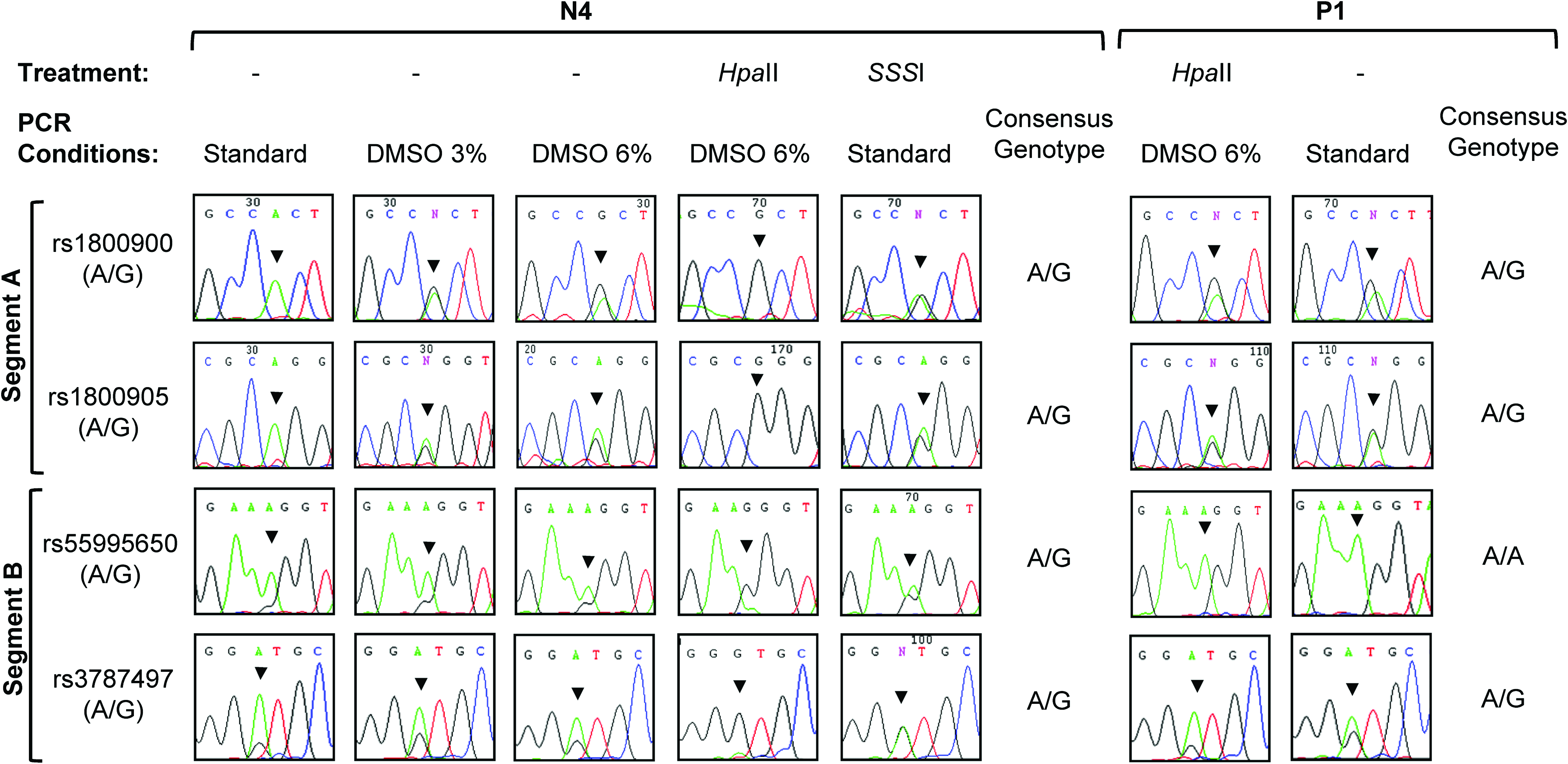
Sequencing analysis of four SNPs at NESP55 DMR. Products A (rs1800900 and rs1800905) and B (rs55995056 and rs3787497) were amplified from normal control 4 (N4) and patient 1 (P1) genomic and HpaII-treated DNA and from N4 SSSI-treated DNA, following standard or optimized PCR conditions, as indicated. Arrowheads indicate the position of the SNP in each electropherogram. Consensus genotypes for each DNA sample are represented. PCR, polymerase chain reaction; SNP, single-nucleotide-polymorphism. Color images available online at www.liebertonline.com/gtmb.
Discussion
We herein report that differential methylation in imprinted genes can lead to PCR bias and allele dropout, which may result in the misgenotyping of heterozygous samples.
For one marker, sequencing-based genotyping, with and without coadjuvant DMSO, was confirmed using BstUI hydrolysis in the 10 control samples. Taking this into account, the misdetection of heterozygotes is not likely to be the result of automated sequencing inaccuracies (Simsek et al., 2001), but it is rather due to a biased PCR amplification. In all amplifications of genomic DNA using standard conditions, the sequencing of control samples yielded a smaller (or a missing) secondary peak at each SNP loci. The less-amplified peaks always corresponded to the methylated allele, as confirmed by sequencing products amplified from HpaII-digested DNA. Haplotypes and genotypes from each normal control were independently confirmed by sequencing, after the insertion of a PCR product (covering all SNPs) into a vector. These PCR products were amplified using independent PCR primers, reagents, and coadjuvants. In the DNA from one PHP-Ib patient, who carried methylation at NESP55 DMR, for all heterozygous SNPs both alleles were always equally amplified with standard conditions. These results were reproduced using an in vitro methylated control DNA sample. These results showed that amplification is not prevented by methylation, but it is rather driven preferentially toward the unmethylated allele when the methylation status of the two alleles is distinct.
CpG methylation has been suggested to alter the DNA secondary structure and melting properties (Laprise and Gray, 2007), which may impair PCR amplification (Kholod et al., 2007). However, to our knowledge, the interference of the differential methylation at imprinted regions in PCR amplification has never been reported. DMSO is a denaturating agent, and the addition of this coadjuvant to the PCR lowers the melting temperature of DNA and facilitates proper primer annealing, allowing amplification of highly methylated regions (Kholod et al., 2007). We used different concentrations of DMSO in the PCR and showed that a concentration of 6% (v/v) was sufficient to prevent allele dropout at NESP55 DMR, possibly by facilitating a balanced denaturation of methylated and unmethylated DNA sequences.
PCR is a well-established and essential tool in the diagnostic medicine, where reliability of results is mandatory. To avoid allele dropout and to ensure high accuracy in genotyping and mutation screening in families with imprinting syndromes, the use of coadjuvants that improve DNA denaturation in the PCRs is highly recommended.
Footnotes
Acknowledgment
This work was funded by Associação de Endocrinologia Oncológica.
Disclosure Statement
No competing financial interests exist.