
Abstract
The identities and frequencies of MYO15A mutations associated with hearing loss in different populations remained largely unknown. We screened the MYO15A gene for mutations in 104 unrelated multiplex and consanguineous Turkish families with autosomal recessive nonsyndromic sensorineural hearing loss using autozygosity mapping. The screening of MYO15A in 10 families mapped to the DFNB3 locus revealed five previously unreported mutations: p.Y289X (1 family), p.V1400M (1 family), p.S1481P (1 family), p.R1937TfsX10 (3 families), and p.S3335AfsX121 (2 families). Recurrent mutations were associated with conserved haplotypes suggesting the presence of founder effects. Severe to profound sensorineural hearing loss was observed in all subjects with homozygous mutations except for two members of a family who were homozygous for the p.Y289X mutation in the N-terminal extension domain and had considerable residual hearing. We estimate the prevalence of homozygous MYO15A mutations in autosomal recessive nonsyndromic deafness in Turkey as 0.062 (95% confidence interval is 0.020-0.105).
Introduction
C
Myosin proteins play crucial roles for normal auditory function. Myosins are actin-dependent molecular motors and use adenosine triphosphate (ATP) to generate mechanical force. They have roles in cell migration and adhesion, intracellular transport, organelle and macromolecule localization, and signal transfer (Probst et al., 1998; Anderson et al., 2000). The myosin superfamily is grouped into 1 conventional and 20 unconventional classes. These classes are divided based on their amino acid diversity of the motor domain (Berg et al., 2001; Krendel and Mooseker, 2005). Mutations in the genes coding for unconventional myosins Ia, IIIa, VI, VIIa, and XVa and conventional myosin heavy chains 9 and 14 have been shown to cause sensorineural hearing loss (Avraham et al., 1995; Friedman et al., 1995; Probst et al., 1998; Wang et al., 1998; Liang et al., 1999; Kalay et al., 2007). Myosin molecules have three main regions (head, neck, and tail). The head region includes an N-terminal extension and motor domains. The N-terminal domain has roles for actin binding and ATP hydrolysis. The motor domain is responsible for ATP activity. The neck region of the protein comprises IQ motifs where calmodulin light chains bind. The tail region includes MyTH4 (myosin tail homolog 4), FERM (band 4.1 erin/radixin/moesin), and SH3 (scr-homology-3) domains and ITLL (class I PDZ-ligand) at the C terminal. Myosin XVa interacts with whirlin through the C-terminal PDZ domain (Belyantseva et al., 2005; Krendel and Mooseker, 2005; Shearer et al., 2009). MYO15A has 66 coding exons that encode a 3,530 amino acid protein. Myosin XVa has important roles for differentiation and elongation of the inner ear hair cell stereocilia (Abecasis et al., 2001; Belyantseva et al., 2003; Krendel and Mooseker, 2005), and is necessary for actin organization in hair cells (Wang et al., 1998; Berg et al., 2001).
The DFNB3 locus was first mapped to the 17p11.2 chromosome region in a population in Bali (Friedman et al., 1995), and later biallelic mutations in MYO15A were shown to be associated with congenital nonsyndromic profound sensorineural hearing loss in humans and in the shaker-2 mouse (Probst et al., 1998; Wang et al., 1998). However, there is little translation of the already available scientific knowledge on MYO15A into clinical practice, because diagnostic testing is not offered due to its large size. Thus, the frequency and spectrum of MYO15A mutations in different populations have remained largely unknown. If a limited number of mutations is common in a population, an initial screening for those mutations can be offered to cases from that ancestry. The identification of recurrent mutations across populations is also important to understanding the genealogy of the mutations and their relationship with historical human movements. To find MYO15A mutations that are associated with nonsyndromic autosomal recessive deafness in Turkey, we studied a large series of families with sensorineural hearing loss.
Materials and Methods
Screened families
We screened 104 unrelated Turkish families in which at least two children were born to consanguineous parents with congenital or prelingual onset nonsyndromic severe to profound sensorineural hearing loss. We considered that hearing loss was a monogenic autosomal recessive trait in each family. The study was approved by the Ankara University School of Medicine Ethics Committee. A signed informed consent was obtained form each participant or parent before the study. Each affected person was evaluated with a standardized history and physical examination protocol that included specific inquries about environmental causes, including aminoglycoside exposure, and full clinical genetics and ENT examination. Sensorineural hearing loss was diagnosed by audiologists using standard audiometry or brainstem auditory-evoked responses. Briefly, pure tone hearing thresholds were determined between 125 and 8000 Hz, according to ISO-1964 standards. Hearing thresholds and tests of speech perception were made in the sound-proof rooms of Industrial Acoustic Company, Inc. with Interacoustics AC-40 audiometry. Air conduction hearing thresholds were measured between 125 and 8000 Hz using TDH-39 standard earphones. Bone conduction hearing thresholds were measured between 0.5 and 4 kHz using Oticon 60273 vibrators. The speech reception threshold was performed using a three-syllable word list, and the speech recognition was performed using a monosyllable phonetically balanced word list. The uncomfortable loudness level was also determined. For immitancemetric evaluation, middle ear pressure and acoustic reflex measurements were made, using Interacoustics AT-22 impedancemeter and TDH-39 earphones. Pure tone average of air conduction thresholds at 500, 1000, and 2000 Hz were used for classification of the severity of hearing loss: normal hearing, <26 dB hearing level (HL); mild hearing loss, 26-40 dB HL; moderate hearing loss, 41-55 dB HL; moderately severe hearing loss, 56-70 dB HL; severe hearing loss, 71-90 dB HL; and profound hearing loss, >90 dB HL.
The physical examination included a thorough assessment with special attention to syndromic findings. All affected subjects were evaluated for the presence of retinitis pigmentosa or other eye findings. An electrocardiogram was obtained in deaf subjects for evaluation of the QT interval for the diagnosis of Jervell and Lange-Nielsen syndrome. All affected subjects were questioned and examined for vestibular findings.
Genomic DNA was extracted from peripheral blood using a standard phenol-chloroform method. Mutations in the noncoding and coding exons of GJB2 as well as the A1555G mutation in the mitochondrial DNA were prescreened in the probands of all families and found to be negative
Locus assignment
At least two affected siblings in 52 families, which included at least three affected children, were screened with GeneChip 10K Xba 142 2.0 Array and Assay kits according to the manufacturer's instructions (Affymetrix, Maumee, OH). This chip contains 10,200 single-nucleotide polymorphisms (SNPs) with a mean intermarker distance of 258 kb, equivalent to 0.36 cM. Briefly, a total of 250 ng intact genomic DNA was digested with XbaI, adaptor ligated, polymerase chain reaction (PCR) amplified, purified, fragmented, and labeled. Cel files obtained from GCOS (GeneChip Operating Software) were processed to CHP file on GTYPE software. Genotypes called by GTYPE software were exported in a table format. For quality control, genotypes exported from GTYPE software were visually evaluated for heterozygosity on chromosome X in males. Family relationship errors were evaluated using the GRR (Graphical Relationship Representation) program (Abecasis et al., 2001). For detection of Mendelian errors, the PedCheck program was used (O'Connell and Weeks, 1998). Genotype data were transferred into a Microsoft Excel sheet and sorted according to the genomic localization of the genotyped SNPs (NCBI Build 36.1; hg18 accessed through the USCS genome browser).
Microsatellite markers D17S1857 (16,355,942-16,356,318 bp), D17S2196 (17,205,118-17,205,398 bp), D17S620 (17,520,397-17,520,859 bp), D17S805 (19,216,034-19,416,333 bp), and D17S689 (20,903,392-20,903,684 bp) flanking the MYO15A gene (17,962,621-18,023,841 bp) were genotyped to evaluate homozygosity of a haplotype in the affected members of the remaining 52 families who had two affected children. PCRs for microsatellite typing were optimized using touchdown programs. After PCR, products were denatured at 95°C with addition of a loading buffer, and were loaded on to 6% denaturing polyacrylamide gels in a long vertical electrophoresis apparatus along with an appropriate ladder for sizing (ΦX174 DNA-HaeIII Digest [New England Biolabs, Ipswich, MA]). The products were observed using silver staining. Allele sizes were determined by running selected samples on an automated capillary sequencer (ABI 3130xl; Applied Biosystems, Foster City, CA). When all affected members in a family showed homozygosity for the same flanking SNP or microsatellite markers, the abovementioned microsatellite markers were genotyped in all available members in the families to evaluate cosegregation of the autozygous haplotype with the phenotype.
Multipoint logarithm of the odds (LOD) scores were calculated using GeneHunter v2.1r5 (Kruglyak et al., 1996) and microsatellite genotypes. Fully penetrant autosomal recessive inheritance was chosen with a disease allele frequency of 0.001. The Marshfield database was used for the mapping data of studied markers.
Mutation analysis
Mutation analysis in MYO15A was performed when the DFNB3 locus cosegregated with autosomal recessive deafness in a family. PCR single-strand conformation polymorphism (SSCP) method was used to screen mutations in the MYO15A (NM_016239) gene. All 66 exons and intron-exon boundaries were amplified in two affected members of each family using intronic primers that have been reported previously (Nal et al., 2007). PCR products were mixed with both forward and reverse oligoprimers in an optimized ratio to improve the sensitivity of SSCP by reducing DNA reannealing, excluding interference from intermediate banding patterns caused by excessive unincorporated primers and enhancing the reproducibility of mutation screening (Zhu et al., 2006). To increase the sensitivity, gels were run at two different temperatures: (1) at 4°C and (2) at a temperature that was calculated using the Ts formula of the PCR product (Li et al., 2003). The PCR products were later denatured at 99°C for 7 min after addition of a loading dye, loaded on 7% nondenaturing polyacrylamide gels, and run at 130 V for 18-20 h. To obtain a stable gel temperature, the DeCode (BioRad, Hercules, CA) mutation detection system was used. Results were observed using silver staining.
The PCR products of the samples that showed band changes in SSCP were purified using a MinElute PCR Purification Kit (Qiagen, Hilden, Germany) and sequenced using a dye terminator cycle sequencing kit (Beckman Coulter, Inc., Fullerton, CA). A CEQ 2000XL automated sequencer (Beckman Coulter, Inc.) was used for DNA sequencing. Each detected MYO15A mutation was screened in 100 Turkish control subjects using SSCP.
In silico analysis of detected missense mutations
The effects of missense mutations identified in this study were assessed using two in silico analyses. (1) The ConSeq (http://conseq.tau.ac.il/) is a Web server for the identification of structurally and functionally important residues in protein sequences. ConSeq scaling procedure shows grades from 1 to 9, where 9 signifies conserved and 1 signifies variable residues. (2) SIFT (http://sift.jcvi.org/) is a sequence-homology-based tool that sorts intolerant from tolerant amino acid substitutions and predicts whether an amino acid substitution in a protein will have a phenotypic effect.
For molecular modeling of myosin XVa (NP_057323), the wild-type and mutant amino acid sequences were entered as input to SWISS-Model workspace (Kiefer et al., 2009) to build 3D-models to identify the location of the mutations and characterize possible effects on protein structure and function. The template was identified by the server. The models generated were submitted to the JOY server (Mizuguchi et al., 1998) to characterize at the residue level the secondary structure and solvent accessibility of the structural models and predict possible effects of the mutations in the protein structure and protein function.
Results
Autozygosity mapping revealed 10 families mapped to the DFNB3 locus
Screening with SNP markers in families with three or more affected children showed concordant homozygous haplotypes flanking the MYO15A gene in the affected members of six families (550, 553, 555, 679, 719, and 780). The affected members of four additional families with two affected children (686, 739, 747, and 762) were homozygous for the regional microsatellite markers. The genotyping of all members of these families showed that autosomal recessive deafness cosegregated with the DFNB3 locus in all 10 families.
Five novel MYO15A mutations were identified in eight families
Mutation screening showed five previously unreported homozygous mutations in different regions of MYO15A in the affected members of eight families (Table 1 and Supplemental Fig. S1., available online at www.liebertonline.com). Each identified homozygous mutation cosegregated with hearing loss in all familes (Fig. 1) and was absent in the Turkish hearing control subjects. No mutation was identified in families 550 and 780, despite multipoint LOD scores of 3.73 and 4.78 were calculated, respectively (Supplemental Fig. S2, available online at www.liebertonline.com).
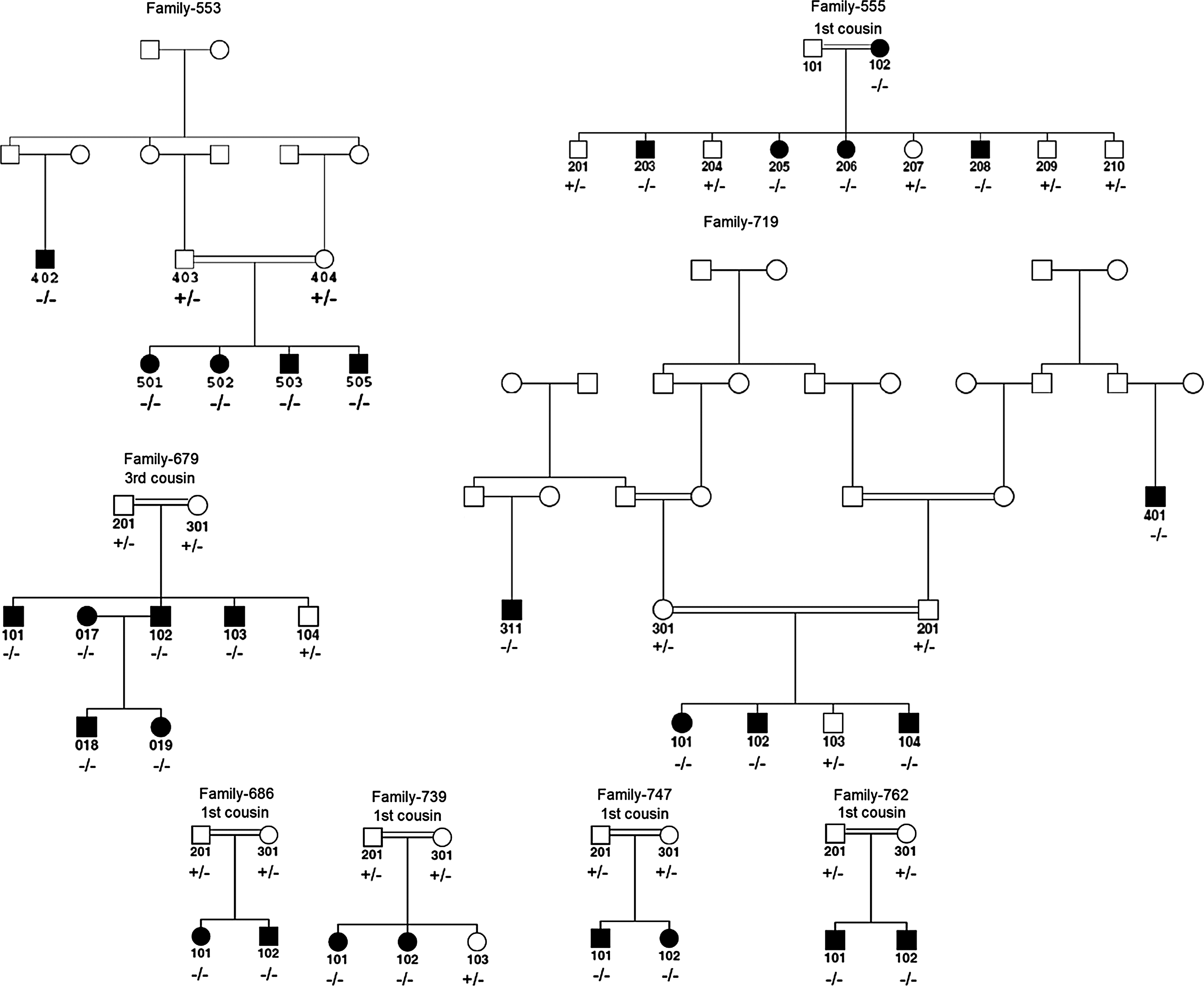
Pedigrees of eight families with MYO15A mutations. Family 553: c.9995_10002dupGCCGGCCC; family 555: p.Y289X; family 679: c.5807_5813delCCCGTGG; family 686: p.S1481P; family 719: c.9995_10002dupGCCGGCCC; family 739: p.V1400M; family 747: c.5807_5813delCCCGTGG; family 762: c.5807_5813delCCCGTGG. −/−: homozygous mutant; +/−: heterozygous; +/+: homozygous wild type genotypes. Consanguinity denied in parents of individuals 402 in family 553; 018 and 019 in family 679; and 311 and 401 in family 719. However, parents in each case originated from the same village. Open and closed squares and circles represent unaffected and affected males and females, respectively.
The hearing phenotype of a homozygous MYO15A mutation suggests a genetic modifier
The hearing phenotype associated with identified homozygous MYO15A mutations was congenital or prelingual onset severe to profound sensorineural hearing loss in all affected individuals except for those in family 555. Individuals 205, 206, and 208 had profound congenital sensorineural hearing loss in this family (Figs. 1 and 2). The mother (102), who was 47 years old, reported that her hearing loss got worse within the past 8 years. An audiogram showed moderately severe sensorineural hearing loss (Fig. 2). Her speech discrimination scores were 48% and 32% in the right and left ears, respectively. Individual 203, who was 28 years old, similarly had some residual hearing and communicated orally. He was not available for the audiological studies. A truncating mutation in the N-terminal extension region of MYO15A, p.Y289X, was homozygous in the mother and all affected children in this family.
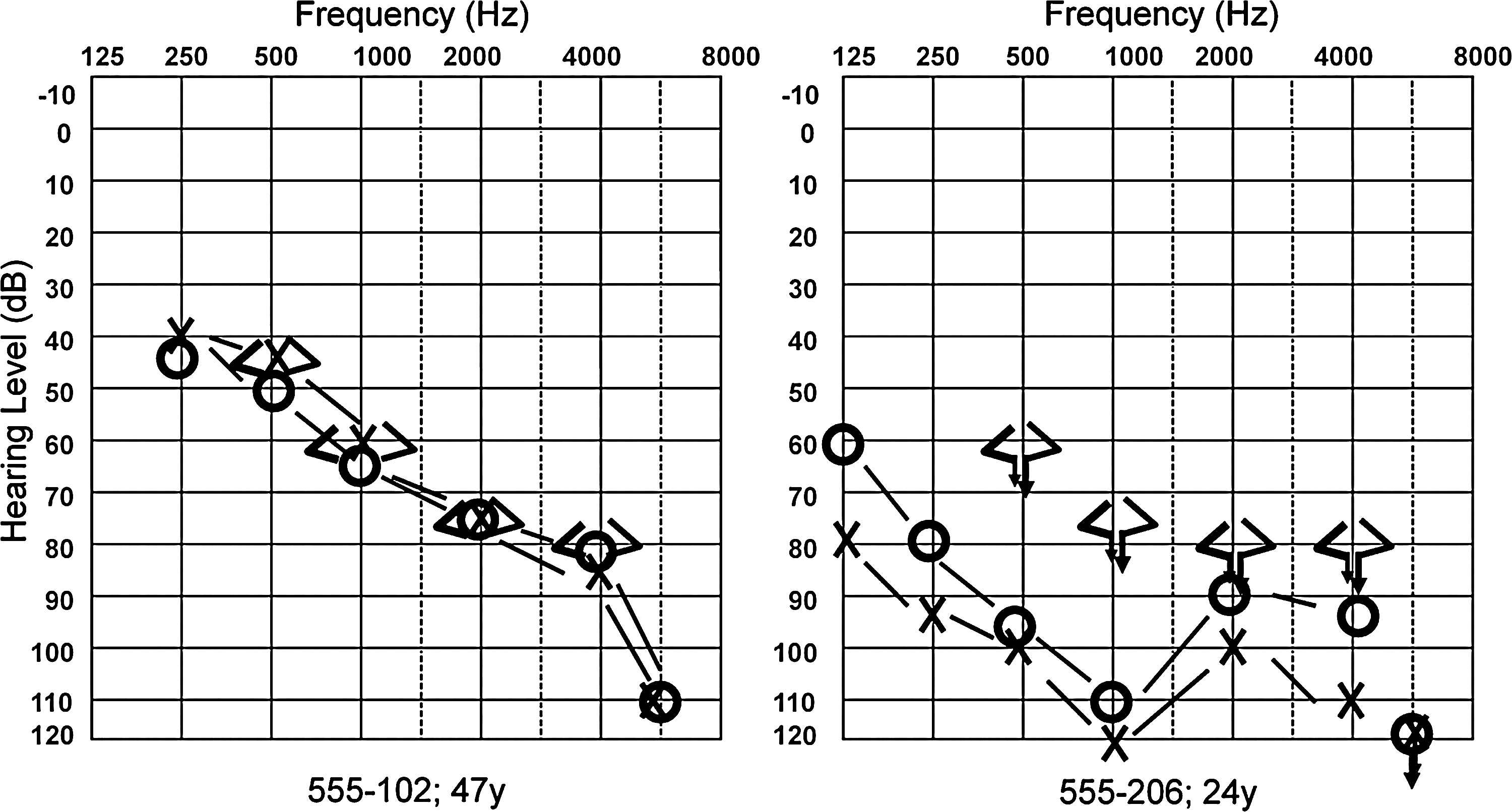
Audiograms of two affected individuals in family 555.
The recurrent MYO15A mutations are associated with conserved haplotypes
Associated haplotypes in families 553 and 719, which had the c.9995_10002dupGCCGGCCC mutation, and in families 679, 747, and 762, which had the c.5807_5813delCCCGTGGG mutation, were conserved and suggested a common origin for each mutation (Table 2).
Heterozygosity rate is according to Caucasians in the Marshfield database. Conserved haplotypes are shaded in gray.
In silico analysis of detected missense mutations (p.V1400M and p.S1481P) supports their pathogenicity
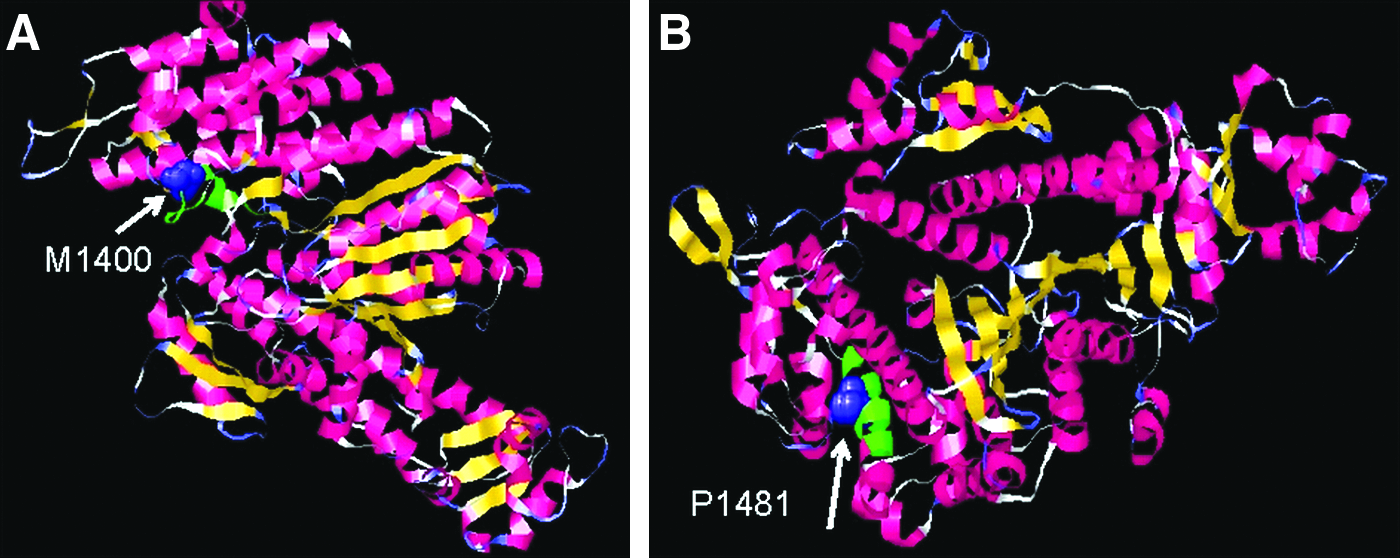
Three molecular models were built using Swiss-Model for the following myosin XVa protein sequences: wild type, p.V1400M, and p.S1481P. The X-ray structure of motor domain of the Dictyostelium Myosin (PDB accession: 1d0x) (Gulick et al., 2000) was identified by the SWISS-MODEL SERVER (Peitsch, 1995; Arnold et al., 2006; Kiefer et al., 2009) as the suitable structural template to base 3D molecular models for the myosin head motor domain in the three myosin XVa protein sequences. The residue range modeled for MYO15_HUMAN (the wild type, p.V1400M, and p.S1481P) was 1220 to1899. For all three models the percent sequence identity ranged between 41.7% and 41.8% (Berman et al., 2000; Gulick et al., 2000) (Fig. 3 and Supplemental Fig. S3, available online at www.liebertonline.com).
(
The local environments of the mutations in the protein structure are shown in Figure 3. V1400 is the residue immediately after helix 12 in a loop and the side-chain solvent accessibility is >7%, while S1481 lies in helix 17 and the side chain accessibility is ≤7% (Supplemental Fig. S3). Using the PFAM sequence alignment (PFAM PF00063), which includes over 1,200 protein sequences from many eukaryote species, at the equivalent position 1400, the following substitutions are observed; T, I, L, C, V, H, A, E, S, G, N, and Y. The ConSeq server shows that the valine residue is highly conserved and the score is 8. Methionine does not occur at this position for the members of this superfamily available in public sequence databases to date. Valine to methionine is a nonconservative substitution. Methionine is an uncommon nonpolar amino acid residue and unlike valine, the side chain is nonbranched and contains a sulphur atom. It is positioned at a solvent accessible site, and we speculate that it may interfere with possible molecular recognition process and indirectly affect the protein function. Finally, SIFT predicts that valine-to-methionine change at position 1400 is not tolerated. In position 1481, we observe S, A, C, G, I, L, and E. The ConSeq conservation score of this residue is 5. Like the last mutation at position 1400, P is not observed at the equivalent position of 1481 in superfamily proteins sequenced to date. Serine mutating to a proline is a nonconservative substitution. This proline is positioned in the middle of a solvent inaccessible helix, and this may disrupt the fold as prolines can act as a structural disruptor in the middle of regular secondary structure. Prolines are known to perturb the structure of alpha helices by introducing a kink between the segment preceding and following the proline residue (Thornton et al., 1988). SIFT software predicts that serine-to-proline change at position 1481 is not tolerated.
Discussion
In this study we describe five novel MYO15A mutations that are predicted to lead to two missense and three truncating (one nonsense, one deletion, and one duplication) alterations in the protein. All detected mutations are associated with nonsyndromic deafness as a fully penetrant autosomal recessive trait in eight Turkish families and are absent in control samples.
The c.867C > G (p.Y289X) mutation affects the protein by truncation. This mutation is found in the second exon that encodes a proline-rich domain at the N-terminal that has no obvious sequence homology to other proteins (Sellers, 2000). It has recently been demonstrated that N-terminal is necessary for normal auditory function. Thus far, only two mutations have been described in this domain (Nal et al., 2007), both of which are truncating. This makes one wonder if missense mutations in this domain are associated with normal hearing, lethality, or a different phenotype. We observed significant residual hearing in two individuals with the homozygous p.Y289X mutation in a family, which suggests the presence of a modifier. It is important to identify more cases with mutations in the N-terminal domain to see if hearing loss is milder than that seen in individuals with mutations in other domains of the protein.
The c.4198G > A (p.V1400M) and c.4441T > C (p.S1481P) mutations are in the motor domain of myosin XVa and were identified in single families in this study. The motor domain is important for the diversity of the myosin superfamilies (Mermall et al., 1998; Probst et al., 1998; Liang et al., 1999) and includes many structural elements that are highly conserved (Friedman et al., 1999). Therefore, multiple missense mutations have been reported in the motor region of MYO15A in families with sensorineural hearing loss (Zhu et al., 2006; Kalay et al., 2007; Nal et al., 2007). A truncating mutation, c.5807_5813delCCCGTGGG (p.R1937TfsX10) also in the motor domain, was identified in three unrelated Turkish families in this study.
The c.9995_10002dupGCCGGCCC (p.S3335AfsX121) mutation, found in two families, is in the FERM domain of myosin XVa. FERM domains mediate binding of cytoskeletal proteins to cytoplasmic domains of transmembrane proteins. Some data suggest that MyTH4/FERM domains in myosin XVa are required for localization of the protein to stereocilia tips, although their specific function has not been elucidated (Belyantseva et al., 2005; Shearer et al., 2009). Our mutation is the third one described in this domain.
We have previously shown that biallelic GJB2 mutations are present in 0.189 of comparable families in Turkey (Tekin and Arici, 2007). Therefore, we estimate the prevalence of homozygous MYO15A mutations in autosomal recessive nonsyndromic deafness in Turkey as 0.062 (95% confidence interval, 0.020-0.105). Considering the extreme locus heterogeneity of deafness, this prevalence is quite significant. Although their deafness was mapped to the DFNB3 locus including MYO15A in two other families (550 and 780), mutation screening remained negative. A formal linkage analysis in these families revealed significant LOD scores. It is possible that changes in the introns or in the regulatory regions of MYO15A might be present in these two families. We may have missed some mutations even in the exons because our screening method was not 100% sensitive. An alternative explanation is the presence of yet another deafness gene at the DFNB3 locus, which has been previously suggested (Nal et al., 2007; Belguith et al., 2009; Shahin et al., 2010).
Haplotype analysis shows that three families having the c.5807_5813delCCCGTGGG mutation and two families with the c.9995_10002dupGCCGGCCC mutation have conserved haplotypes, suggesting a common ancestor for each mutation. These families are from different cities in central Anatolia and deny consanguinity. Turkey has been continually inhabited since ancient times and connected three old continents. The inhabitants of Turkey have traditionally lived in small, relatively isolated villages and are often the descendants of a small number of founders migrating from distant lands. Currently, there are approximately 40,000 villages of this type throughout Turkey. Although migration to urban centers has occurred during the past 50 years, the traditional pattern of consanguineous marriage has persisted, and is currently estimated to involve 20%-25% of all marriages (Tuncbilek, 2001). When a new deafness mutation arises or is introduced with migration into a small and isolated population, genetic drift can lead to a substantial increase in gene frequency in successive generations. When it is accompanied with intense population movements as it has happened in Anatolia throughout human history, gene flow facilitates the spread of the initial founder mutation into different geographic regions. Traditional marriage pattern of inbreeding, resulting either from a small effective population size in a subdivided population or from a consanguineous marriage, in Turkey is highly suitable for phenotypic expression of autosomal recessive deafness. The final consequence is extreme locus and allelic heterogeneity of deafness, with quite a few recurrent mutations reflecting the founder effect and gene flow. The absence of recognized consanguinity between the parents of multiple affected individuals in our families with homozygous mutations illustrates the increased carrier frequency of the detected mutation due to founder effects (Fig. 1). It will not be surprising to discover that MYO15A mutations identified in this study are responsible for hearing loss in other populations.
Footnotes
Acknowledgments
The authors thank the families for participation. This study was supported by The Scientific and Technological Research Council of Turkey (TUBITAK) with a grant (105S464) to M. Tekin.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.