
Abstract
Focal dermal hypoplasia (FDH) is an X-linked dominant disorder caused by mutations in the gene PORCN, which encodes a protein required for the secretion and signaling of Wnt proteins. While deletions are responsible for a small percentage of FDH-causing mutations, the vast majority of mutations are single-nucleotide substitutions or small deletions or insertions that can be identified by sequence analysis. In 2007, we implemented a PORCN gene sequencing test for individuals with a clinical diagnosis of FDH. To date, we have detected 12 novel PORCN mutations and 6 previously reported mutations in 53 such unrelated patients. The pathogenic PORCN mutations included nine nonsense mutations, three missense mutations, one small deletion, two small duplications, and three splice-site mutations. Of these mutations, two were found in affected men and were mosaic; one of these was found in three other affected women. The remaining 16 mutations were found only in women. All the mutations detected in women were presumed heterozygous. In addition to the disease-causing mutations, eight nucleotide variants of unknown significance were identified. Further characterization of these variants suggests that four of them are pathogenic mutations. These findings add to the heterogeneity of mutations in the PORCN gene that cause FDH.
Introduction
F
The gene responsible for FDH was identified as PORCN (Grzeschik et al., 2007; Wang et al., 2007), which encodes an enzyme involved in the secretion of Wnt proteins (Caricasole et al., 2002). Approximately 80% of FDH-causing mutations in PORCN are single-nucleotide substitutions, small deletions, or small insertions identifiable by sequence analysis of the PORCN coding region, while the remaining mutations are large genomic deletions that include PORCN (Grzeschik et al., 2007; Wang et al., 2007; Clements et al., 2008; Leoyklang et al., 2008; Bornholdt et al., 2009; Froyen et al., 2009; Harmsen et al., 2009; Maas et al., 2009); more recently, an intragenic deletion of four exons was identified in a female patient (Bornholdt et al., 2009). Ninety percent of affected individuals are women (Gorlin et al., 2001). Men with FDH are rare; all were found to have somatic mosaicism for mutations in PORCN and were generally more mildly affected than women (Grzeschik et al., 2007; Wang et al., 2007; Bornholdt et al., 2009; Maas et al., 2009). Nonmosaic, hemizygous mutations in men are presumed to cause lethality. Ninety-five percent of all cases and 100% of male cases appear de novo without known male-to-male transmission (Gorlin et al., 2001).
We implemented clinical diagnostic sequencing for PORCN mutation analysis in September 2007 and thus far have studied 53 unrelated probands that were referred with a clinical diagnosis of FDH. Our data herein describe the PORCN mutations identified through sequencing of the gene, and further examine the possible effects of the unclassified variants.
Materials and Methods
Clinical samples
Probands from 53 unrelated families were studied; all patients were referred to the Medical Genetics Laboratories at Baylor College of Medicine for the evaluation of PORCN gene mutations. The reason for referral was a possible diagnosis of FDH. We have limited information on the phenotypes of these patients. All research procedures were performed in compliance with appropriate federal regulations and with the approval of the Baylor College of Medicine Institutional Review Board.
Mutation analysis
Genomic DNA was isolated from blood leukocytes on the AutoPure LS robotic workstation (Qiagen) using the Puregene DNA isolation kit (Qiagen). Polymerase chain reaction (PCR) primers were designed to cover the 14 coding exons and flanking regions of the PORCN gene (NM_203475). After amplification, direct sequence analysis of the PCR products was performed in both forward and reverse directions using automated fluorescence dideoxy sequencing methods and the 3730xl DNA analyzer (Applied Biosystems). All detected mutations and variants were re-analyzed for confirmation.
In silico analysis
Splice prediction programs, namely, the one by Neural Network, NNSPLICE 0.9 version (www.fruitfly.org), and NetGene2 (www.cbs.dtu.dk/services/NetGene2/), were used to predict the splicing consequences of the normal gene sequence versus that of the two splice-site variants, c.555+4A>G and c.374-15G>A. Other computational algorithms, namely, PolyPhen (http://genetics.bwh.harvard.edu/pph/) and SIFT (http://sift.jcvi.org/), were used to predict the functional impact of the de novo c.1021C>T (p.H341Y) mutation and the two missense variants, c.1040T>C (p.L347P) and c.1106C>T (p.A369V).
Reverse transcription-PCR analysis for the c.555+4A>G unclassified variant
For endogenous PORCN RNA splicing assays, total RNA was isolated using the RNeasy kit (Qiagen) from lymphoblastoid cell lines of normal subjects and patients carrying the c.555+4 A>G mutation. Reverse transcription (RT) was performed using the SuperScript III First-Strand DNA Synthesis kit (Invitrogen). Three RT-PCRs were performed to detect the predicted alternatively spliced transcript. The first PCR was performed with primer set 1, which is located in exon 5 (5′-TGGAGTTCATGGGCTACCTC-3′) and exon 8 (5′-TGCTGAAGTGGAAGGAGACA-3′), to amplify both wild-type and the predicted alternatively spliced transcript. The other two PCRs were performed with primer sets 2 and 3 in intron 5 and exon 8 to specifically amplify the predicted alternatively spliced transcript (primer set 2: forward 5′-CACTGGTGGGGTCCTGAGT-3′, reverse 5′-TGCTGAAGTGGAAGGAGACA-3′; primer set 3: forward 5′-CTGGTGGGGTCCTGAGTG-3′, reverse 5′-TGCTGAAGTGGAAGGAGACA-3′; Supplemental Fig. S1, available online at www.liebertonline.com).
Results
Pathogenic mutations
PCR amplification and sequencing of the PORCN coding region was carried out for all 53 unrelated probands with a possible clinical diagnosis of FDH. Disease-causing mutations were identified in two out of six men and 21 out of 47 women with an overall detection rate of ∼43%. Mutations identified in these patients included 6 previously described mutations and 12 novel frameshift, nonsense, missense, or splice-site mutations (Table 1). The mutations in the affected men were found to be mosaic. All mutations detected in women were presumed heterozygous, except for one woman who was mosaic. Among these mutations, c.1021C>T (p.H341Y) and c.1317G>A (p.W439X) were the only mutations confirmed to be de novo in these female patients (patient 12 and patient 20; Table 1) where parental samples were available for confirmation. In silico analysis with PolyPhen and SIFT also showed that c.1021C>T (p.H341Y) was possibly deleterious. For all other patients, either one or both parents were not available.
Wang et al. (2007).
Bornholdt et al. (2009).
Maas et al. (2009).
Leoyklang et al. (2008).
Froyen et al. (2009).
Harmsen et al. (2009).
NA, not applicable.
Characterization of variants of unknown significance
We identified eight novel variants in our cohort of patients, including two missense and six intronic changes (Table 2). In silico analysis with PolyPhen and SIFT showed that both missense changes, c.1040T>C (p.L347P) and c.1106C>T (p.A369V), were possibly deleterious.
Pathogenic mutation also identified (see Table 1).
Predicted to be deleterious with in silico analysis.
Previously reported in the same patient with focal dermal hypoplasia (Schaffer et al., 2009).
NA, not applicable.
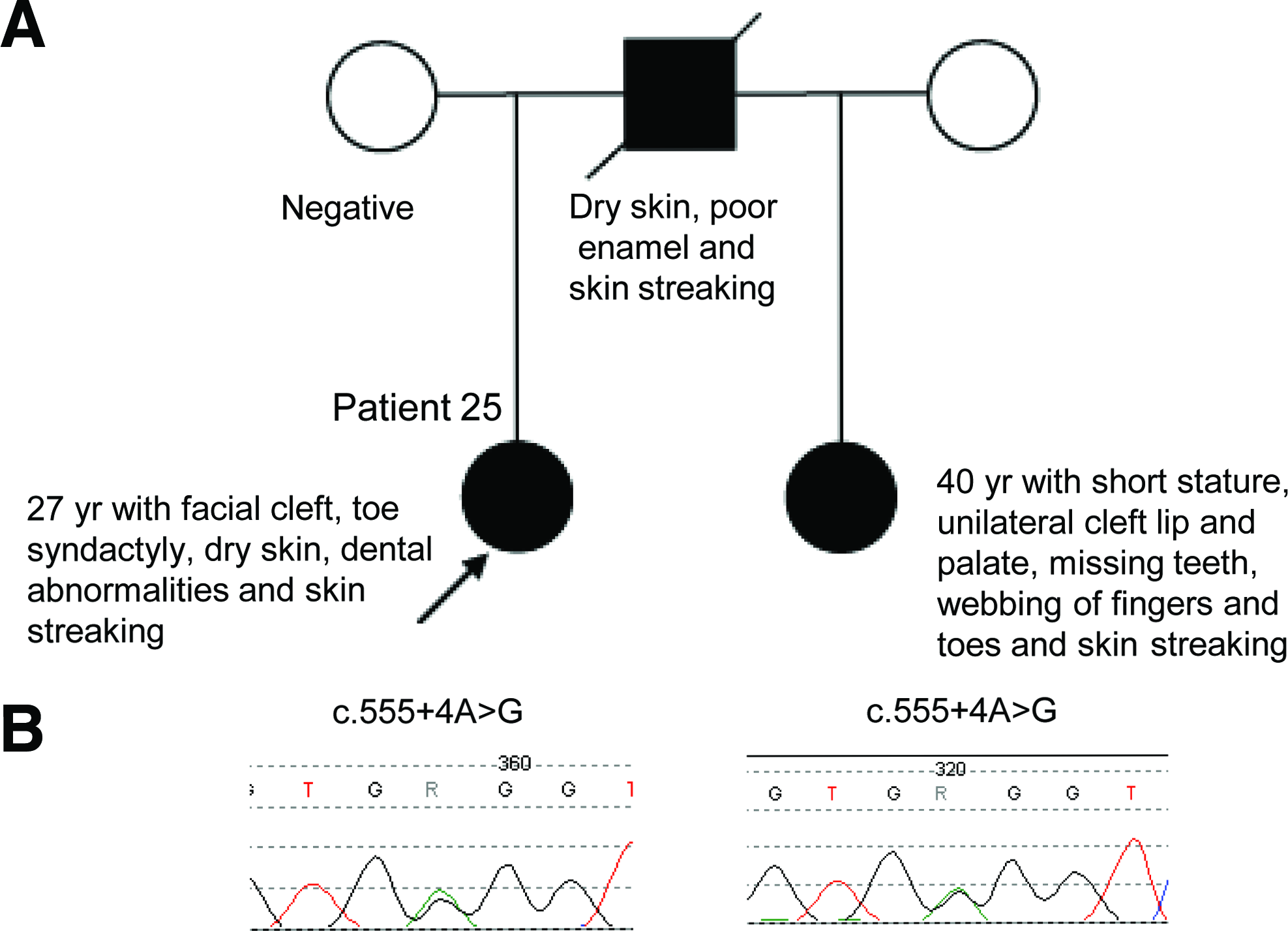
Among the intronic variants, the c.555+4A>G was noted for its inheritance pattern. The c.555+4 A>G variant was identified in female patient 25 and her paternal half-sister (Fig. 1A, B). Both half-sisters presented with classic symptoms of FDH. They appeared to have inherited this variant from their father who reportedly had mild clinical features of FDH, including dry skin, poor enamel, and skin streaking; however, he died from unrelated causes and his DNA sample was unavailable for confirmation. In silico analysis using NNSPLICE and NetGene2 predicted that the c.555+4A>G change reduces the efficiency of the intron 5 splice donor site. It is presumed that an alternatively spliced transcript is formed that includes some nucleotides of intron 5 that contains a premature stop codon, thus suggesting this change to be possibly deleterious. However, RT-PCR using several primer sets and mRNA extracted from the lymphoblastoid cell lines did not result in the identification of alternatively spliced transcripts (data not shown). Additionally, the sequence of the RT-PCR product from the affected patients was not found to contain the alternatively spliced transcript (data not shown). In silico analysis using NetGene2 showed that the c.374-15G>A variant in patient 24 creates a novel acceptor splice site in the sequence, suggesting that this variant is pathogenic. However, NNSPLICE in silico analysis of the same variant did not predict any difference. When in silico analysis was performed with the c.1284+32C>A change in intron 14 seen in patient 28, it did not reveal any difference as compared to the wild-type sequence. Moreover, this sequence change was found in the clinically normal male parent of the proband, indicating that c.1284+32C>A change is benign and not associated with FDH. The remaining intronic variants, c.330-11T>A (patient 22), c.330-27delG (patient 11), and c.720-28C>T (patient 5), were identified in patients who already harbored a pathogenic mutation. Thus, these three variants likely represent benign changes (Table 2).
Discussion
In this report, we identified 12 novel PORCN mutations. These mutations are predicted to result in abnormal splicing, protein truncation, or nonsense-mediated mRNA decay with consequent disrupted PORCN function.
Two PORCN mutations reported previously, c.178G>A (p.G60R) and c.1094G>A (p.R365Q), were found to be recurrent in this study (Wang et al., 2007; Leoyklang et al., 2008). The p.G60R (c.178G>A) was reported previously as a mutation in one female patient (Wang et al., 2007) and was identified in three out of 53 unrelated female probands in our cohort. The c.1094G>A (p.R365Q) was identified previously as a mutation in four female patients (Leoyklang et al., 2008; Bornholdt et al., 2009; Froyen et al., 2009; Maas et al., 2009). Our diagnostic study detected this mutation in an additional 4 out of 53 unrelated probands. Combining previous studies and the current study, the c.1094G>A (p.R365Q) mutation was not detected in at least 680 control X chromosomes (Leoyklang et al., 2008; Bornholdt et al., 2009; Froyen et al., 2009; Maas et al., 2009). Interestingly, each of these nucleotides, c.178G and c.1094G, is part of a CpG dinucleotide and is therefore likely to represent a hotspot for PORCN gene mutations.
The c.1021C>T (p.H341Y) change represents a disease-causing mutation in patient 12 due to its de novo nature. Additionally, molecular modeling studies have predicted histidine 341 to be the active site of PORCN (Bornholdt et al., 2009), suggesting that any substitution of this histidine residue would be deleterious. This is in agreement with the c.1022A>T (p.H341L) change that was previously reported as a de novo mutation in a female patient with FDH (Bornholdt et al., 2009).
The two interesting missense variants that warrant further functional investigations are c.1040T>C (p.L347P) and c.1106C>T (p.A369V). Molecular modeling studies suggest that the L347 residue lies in the PORCN transmembrane domain and the A369 residue lies in the luminal side of the PORCN protein, and that both these domains contribute to the functionality of PORCN (Bornholdt et al., 2009).
By in silico analysis the c.555+4A>G variant was predicted to create an alternative-spliced transcript. However, our RT-PCR analysis did not reveal the predicted abnormal splice variant, strongly suggesting nonsense-mediated RNA decay of the mutant transcript.
Patient 24 with the c.374-15G>A change was previously reported by Schaffer et al. (2009). The nucleotide variation in this patient was identified in our laboratory and interpreted as a variant of unknown clinical significance in the diagnostic report. However, this variant was interpreted by Schaffer et al. (2009) to be a pathogenic mutation, based on clinical findings of classical features of FDH along with syringocystadenoma papilliferum. There are no functional studies at the transcriptional level to support the deleterious nature of this variant, and no other unrelated FDH patients have been reported to carry this variant. This variant should be classified as a variant with unknown clinical significance until additional experimental data is available.
A summary of all the mutations identified to date through sequence analysis and microdeletion testing is presented in Figure 2. Among those that can be identified through sequence analysis, single-nucleotide substitutions comprised of missense, nonsense, and splice-site mutations constitute ∼67% of the mutations, while small deletions and insertions leading to frameshifts account for the remainder mutations (Fig. 2). One prior study identified a deletion of exons 1 through 4 in a patient with FDH (Bornholdt et al., 2009). These data therefore provide evidence for the existence of intragenic deletions of PORCN. Thus, it would be worthwhile to investigate patients with FDH who have no mutations by DNA sequencing for such intragenic deletions through the use of a targeted high-density genomic array, multiplex ligation-dependent probe amplification (MLPA)/multiplex amplifiable probe hybridization (MAPH) analysis, or Southern analysis.
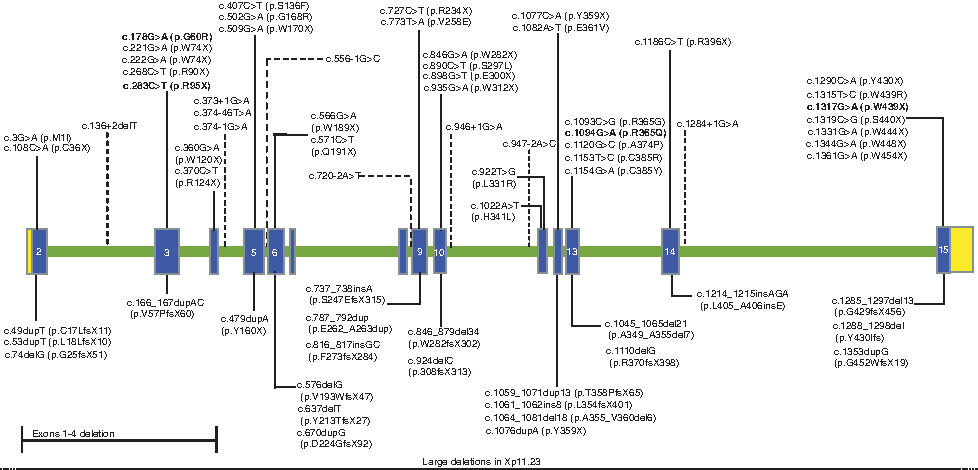
Classification of mutations identified in patients with focal dermal hypoplasia through sequence analysis of the PORCN gene (NM_203475). The blue boxes indicate the coding regions of the gene (exon 2 through exon 15), the green bar represents the intronic region, while the yellow boxes indicate the 5′ and the 3′ untranslated regions of the gene. The mutations above the gene are single-nucleotide substitutions that include missense, nonsense, and splice-site mutations (indicated with dotted lines), whereas the mutations below the gene are large deletions and frameshift mutations due to small duplications or deletions. Previously identified mutations (Grzeschik et al., 2007; Wang et al., 2007; Clements et al., 2008, 2009; Leoyklang et al., 2008; Bornholdt et al., 2009; Froyen et al., 2009; Harmsen et al., 2009; Maas et al., 2009) and those newly identified in this study are shown in black, and the recurrent mutations are in bold. Color images available online at www.liebertonline.com/gtmb.
The spectrum of mutations identified to date consists mainly of small mutations within the PORCN gene as well as large deletions (Grzeschik et al., 2007; Wang et al., 2007; Clements et al., 2008, 2009; Leoyklang et al., 2008; Bornholdt et al., 2009; Froyen et al., 2009; Harmsen et al., 2009; Maas et al., 2009). In review of the published literature on 92 patients with clinical diagnosis of FDH, 79 patients had detectable PORCN mutations, which brings the overall detection rate to ∼86% (Grzeschik et al., 2007; Wang et al., 2007; Bornholdt et al., 2009; Froyen et al., 2009; Harmsen et al., 2009; Maas et al., 2009). Seventy-five percent (69/92) of these patients had small mutations that were detected by sequence analysis, while large deletions detected by array comparative genomic hybridization (CGH), qPCR, and Multiplex Amplifiable Probe Hybridization (MAPH) constituted about ∼11% (10/92). Our sequence base analysis has found an overall detection rate of ∼47% for small mutations, which is 28% lower than the published data. This detection rate reflects the nature of a routine clinical diagnostic laboratory setting wherein patients with a broad range of clinical presentation may be referred for testing and some may not fulfill a clinical diagnosis of FDH. Our detection rate therefore should not be interpreted as the true mutation rate in classic and clinically confirmed FDH.
In conclusion, we report 12 novel mutations and 6 mutations previously described in our cohort of 53 unrelated probands whose DNA was sent to our laboratory for clinical diagnostic testing. This study also identifies the variants of unknown significance in our patients and their predicted outcome through in silico analyses. Overall, these findings will contribute to the expanding literature of PORCN mutations.
Footnotes
Acknowledgments
This work was funded by the March of Dimes and the National Foundation for Ectodermal Dysplasias.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.