
Abstract
Janus Kinase 2 (JAK2) is a member of a family of four Janus Kinases, 2, and 3 and tyrosine kinase 2. Mutated JAK2 (V617F) has the ability to activate downstream signal transducer and activator of transcription (STAT)-mediated transcription in the absence of the ligand erythropoietin. The autoinhibitory activity of JAK2 is disrupted by the presence of the V617F mutation. Somatic mutation in JAK2 (V617F) gene has been reported in myeloid disorders. This study reports the prevalence of JAK2V617F using amplification refractory mutation system (ARMS)-polymerase chain reaction in 246 Egyptian patients with different myeloid disorders and studied the relationship between the JAK2V617F mutation and parameters in peripheral blood. The mutation was detected among 88 patients (35.8%) with different myeloid disorders. JAK2V617F was found among 81.4% of polycythemia vera (PV), 50% of essential thrombocythemia, 46.1% of primary myelofibrosis (PMF), 33.3% of philadelphia (Ph)-negative chronic myeloid leukemia, 33.3% of myelodysplastic syndrome (MDS)/myeloproliferative neoplasm (MPN), and 50% of refractory anemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T) patients. Hemoglobin and white blood cells were significantly higher in the mutated group of MPN including PV, essential thrombocythemia, and PMF, whereas platelet counts were higher among the mutated PV, PMF, RARS-T, and MDS/MPN group. The identification of JAK2V617F mutations has raised the prospect of developing specific JAK2V617F inhibitors to treat mutated patients.
Introduction
The molecular characterization of PV came in 2005 with the discovery of the Janus Kinase 2 (JAK2) V617F mutation in about 90% of PV patients. While studying the mechanisms responsible for the erythropoietin (EPO)-independent growth characteristic of PV progenitors (Ugo et al., 2004; Baxter et al., 2005; James et al., 2005b; Jelinek et al., 2005), the team of William Vainchenker discovered the presence of a mutated form of the JAK2 protein, JAK2V617F (James et al., 2005b). Experiments performed showed that the mutated JAK2V617F was constitutively active and able to activate the erythropoietin receptor (EPOR) signaling pathway without EPO (James et al., 2005b; Baxter et al., 2005). Indeed, the JAK2V617F mutation was found not only in almost all patients with PV but also in about 50% of patients with ET and PMF, in rare patients with CML and atypical MPN (James et al., 2005a; Jelinek et al., 2005), in myelodysplastic syndromes with thrombocytosis (Schmitt-Graeff et al., 2008), and even in hematologically normal patients with portal vein thrombosis (Kiladjian et al., 2008b).
The JAK2V617F mutation involves a guanine-to-thymine mutation in exon 14 resulting in a substitution of valine to phenylalanine at codon 617 within the pseudokinase domain JH2, which disrupts the autoinhibition of this regulatory domain. Consequently, the tyrosine kinase corresponding to JH1 domain is constitutively activated, which can bind to a cytokine receptor and promote STAT recruitment (James et al., 2005b; Levine et al., 2005).
Expression of JAK2V617F in hematopoietic cells activates intracellular signaling pathways downstream of the EPOR, including the STAT5, STAT3, and mitogen activated protein kinases (MAPK) pathways (Silva et al., 1998; James et al., 2005b). STAT5 is normally phosphorylated by the cytokine receptor-JAK2 complex, and the phosphorylated STAT5 then translocates to the nucleus and activates the transcription of target genes including bcl-xl, an important antiapoptotic protein known to be expressed in high levels in PV proerythroblasts. It is important to determine that coexpression of wild-type JAK2 does not interfere with the ability of JAK2V617F to autophosphorylate, even when wild-type JAK2 is expressed at higher levels than the mutant kinase (Hoffman et al., 2008).
Large differences were observed among MPN patients during quantitative assays for analysis of JAK2V617F mutant allele burdens ranging from 0% to 100% (homozygosity) in PV and PMF, whereas patients with ET carry burdens between 0% and 50% and only rarely exceed 50% (heterozygosity). Low JAK2V617F mutational burden in ET is largely due to the absence of 9p unipaternal disomy in these patients (Silva et al., 1998; Hoffman et al., 2008; Scott et al., 2006).
The less-frequent exon 12 mutations were observed in PV patients only with a frequency of up to 5% (Scott et al., 2007; Pietra et al., 2008). These mutations might define a distinctive myeloproliferative syndrome that affects patients who currently carry a diagnosis of idiopathic erythrocytosis. These patients had reduced serum EPO levels (Martinez-Aviles et al., 2007). Patients with a familial history of MPN are phenotypically and molecularly indistinguishable from sporadic MPN patients (Kralovics et al., 2003; Bellanne-Chantelot et al., 2006; Rumi et al., 2006). All familial MPN cases tested to date were carrying a somatic JAK2V617F or JAK2 exon 12 (Bellanne-Chantelot et al., 2006).
Although myelodysplastic syndrome (MDS) and MPN appear to have entirely different pathophysiological mechanisms, the existence of conditions with overlapping features has been well established. In 2002, Schimtt-Graeff et al. (2002) published a study showing both thrombocytosis in peripheral blood and ringed sideroblasts in the bone marrow, a condition that was at that time defined as ET with ringed sideroblasts. Findings of this study provided evidence that ET with ringed sideroblasts includes a wide spectrum of conditions ranging from MDS to MPN. Several studies on mutation analysis of JAK2V617F have reported the presence of the mutation among MDS/MPN (Szpurka et al., 2006; Schmitt-Graeff et al., 2008).
Our objective in the present study was to confirm the reported prevalence of JAK2V617F in different myeloid disorders, including CML, PV, ET, PMF, AML, MDS, MDS/MPN, and RARS-T, and to assess the relationship between JAK2V617F mutation and parameters in peripheral blood.
Patients and Methods
The study included 246 patients having myeloid disorders, in addition to 20 healthy controls matched for age and sex. Age range was between 10 and 78 years (mean: 50.07 ± 14.5). Diagnosis of myeloid disorders was based on WHO criteria for classification. All patients were selected from those attending the Alexandria main university hospital hematology unit during the period from January 2008 till January 2010.
The study was approved by the Research Ethics Committee of Alexandria University and a written informed consent was obtained from all patients participating in the study.
All patients were subjected to:
Full history evaluation-complete clinical examination:- Complete blood picture (Bain and Bates, 2006)- Bone marrow aspiration and/or biopsy to confirm diagnosis (Bates and Lewis, 2006)- Reticulin stain to confirm the diagnosis of myelofibrosis (Bain and Swirsky, 2006) Cytogenetic studies for the presence of Philadelphia chromosome in MPNs (Wang et al., 2001) JAK2V617F genotyping (Jones et al., 2005)
JAK2V617F genotyping
The JAK2-ARMS assay is a multiplex polymerase chain reaction (PCR) (Ke et al., 2002; Jelinek et al., 2005) that uses two primer pairs to amplify normal and mutant sequences, as well as a primer pair that amplifies a DNA fragment flanking the mutation site. The latter is ubiquitous in all reactions and is used as an internal control in our test assay. Both mutant- and wild-type-specific primers include appropriate mismatches to maximize discrimination between mutant and wild-type alleles, respectively. Based on the genotype of the sample analyzed, the PCR was predicted to yield the following fragments:
For a wild-type genotype, a fragment of 463 bp corresponding to the internal control amplicon and a fragment of 229 bp corresponding to the wild-type amplicon. For a mutant genotype, the ubiquitous 463-bp fragment in addition to the wild-type (229 bp) and the mutant (279 bp) amplicons are detected in heterozygous cases, whereas in homozygous cases only the mutant (279 bp) amplicon is detected.
Genomic DNA was extracted from either peripheral blood (PB) or bone marrow (BM) cells using a GFX genomic blood purification kit (Amersham, Biosciences). Extracted DNA was then amplified by PCR.
Primers used were forward outer (5′-TCC TCA GAA CGT TGA TGG CAG TTG CAG-3′), reverse outer (5′-ATT GCT TTC CTT TTT CAC AAG AT-3′), forward wild-type specific (5′-GCA TTT GGT TTT AAA TTA TGG AGT ATG TG-3′), and reverse mutant specific (5′-GTT TTA CTT ACT CTC GTC TCC ACA GAA-3′).
The total reaction volume of 25 μL contained 250 ng DNA and 20 pmol each primer. The 2 × PCR master mix (Fermentas Life Science) contained 0.05 units/mL of Taq polymerase in reaction buffer.
Amplifications were performed in a DNA thermal cycler (Techne Cambridge) using the following protocol: denaturing step at 95°C for 5 min; 95°C for 30 s, 59°C for 30 s, and 72°C for 1 min, for 30 cycles; final extension: 72°C for 5 min, followed by 4°C. Products were resolved on 2% agarose gels and visualized under ultraviolet illumination after staining with ethidium bromide (Fig. 1).
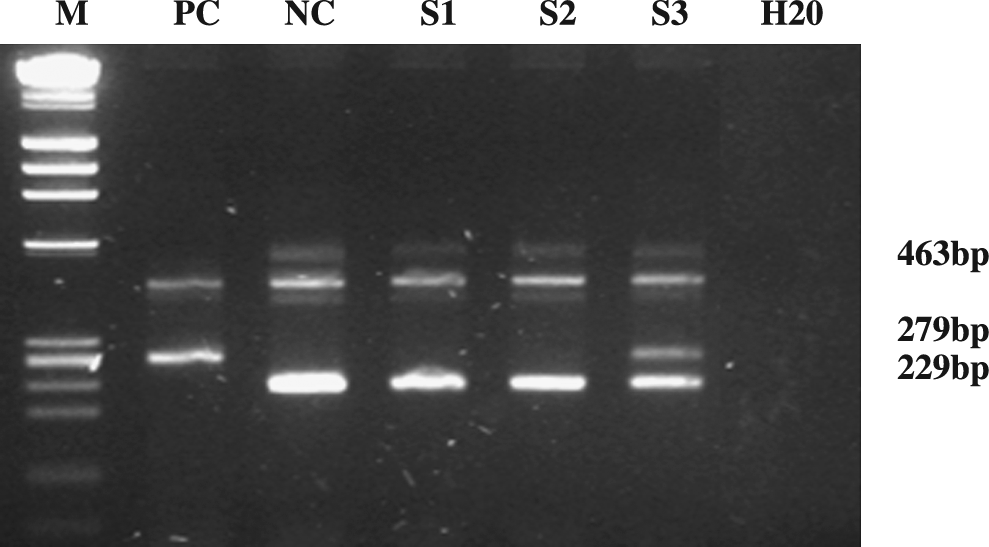
Agarose gel electophoresis. M: molecular marker; PC: homozygous case; NC: wild type; S1, S2: wild-type samples; S3: heterozygous showing both mutant (279 bp) and wild (229 bp) alleles.
Statistical analysis
Data were collected and entered into the personal computer. Statistical analysis was done using Statistical Package for Social Sciences (SPSS, version 15) software.
The following statistical tests were used: arithmetic mean and standard deviation for categorized parameters, chi-square test for numerical data, analysis of variance test for more than two groups, and t-test for two groups. The level of significance was 0.05.
Results
We studied samples from a total of 246 patients with different myeloid disorders for the presence of JAK2V617F exon 14 mutation: 50 chronic myelogenous leukemia (CML) (44 Ph+ and 6 Ph−), 54 PV, 30 ET, 39 PMF, 12 RARS-T, 9MDS, 9 MDS/MPN, and 43 AML patients.
Frequency of JAK2V617F mutation in the studied group
JAK2V617F mutation shows a wide morphologic spectrum. Eighty-eight patients (35.8%) were positive for the mutation, and among them 82 were heterozygous and 6 were homozygous. The mutation was present in 2 Ph− CML (2.2%), 15 ET (17%), 18 PMF (20.4%), 44 PV (50%), 6 RARS-T (6.2%), and 3 MDS/MPN (3.4%) patients. The frequency of the mutation in the different myeloid disorders was heterogeneous and ranged from 0% in AML, Ph+ CML, and MDS to 81.4% in PV. The mutation was significantly higher among PV patients (p < 0.05).
Frequency of JAK2V617F in the different groups
JAK2V617F was found in 81.4% of PV patients, 50% of ET, 46.1% of PMF, 33.3% of MDS/MPN, and 50% of RARS-T, and in Ph− CML the mutation was found in 1 (16.6%) of 6 patients.
Relation between JAK2V617F mutation and parameters in peripheral blood
We found that hemoglobin level was higher in MPN (PV, ET, PMF) positive for JAK2V617F mutation when compared with those negative for JAK2V617F, reaching the significance level in PMF and ET (p = 0.013, p = 0.011; Table 1).
= significant < 0.05.
PV, polycythemia vera; MF, myelofibrosis; ET, essential thrombocythemia; Hb, hemoglobin; RBCs, red blood cells; WBCs, white blood cells; SD, standard deviation.
As regarding white blood cells, it was significantly higher among PV, ET, and PMF patients positive for the JAK2V617F mutation in comparison with unmutated PV, ET, and PMF patients (p = 0.038, p = 0.001, p = 0.01). Platelet counts were significantly higher among mutated than unmutated patients with PV and PMF (p < 0.05). Platelet count was higher in RARS-T and MDS/MPN positive for JAK2V617F mutation, whereas it was significantly lower among JAK2V617F positive ET (p = 0.001).
The mutation was more frequent in females (58.7%) than males (41.3%). There was no age difference among positive and negative patients.
Discussion
The V617F gain-of-function mutation in exon 14 of JAK2 gene has been recognized as the major pathogenetic event in chronic myeloproliferative disorders, particularly PV. This study identified the JAK2V617F mutation in ∼36% of patients with different myeloid disorders as well as confirmed the presence of this mutation in the great majority of patients with PV and nearly half of cases with ET or PMF (85%, 50%, and 46%, respectively) (Jelinek et al., 2005). This intriguing feature raises the major unanswered question of how a unique mutation can cause different phenotypes. Although it is likely that gene dosage for JAK2V617F is an important contributor to MPN phenotype, there are data to suggest that additional genetic and epigenetic factors, including germ-line modifiers and epigenetic silencing of JAK-STAT pathway genes, contribute to MPN pathogenesis. It is also possible that additional somatic mutations, which remain to be identified, play an important role in instructing the phenotype of JAK2V617F-positive hematopoietic progenitors. There are now several arguments that strongly suggest that a preceding event occurs before JAK2V617F in some patients (Kralovics et al., 2006).
We found that ET JAK2V617F-positive patients had higher hemoglobin levels and lower platelet counts, which increased suggestions that these patients more closely resembled PV with increased erythropoiesis and granulopoiesis, whereas JAK2V617F-negative ET patients were characterized by a greater degree of megakaryopoiesis. Such data may be due to different signal transduction pathways or the presence of MPLW515 mutation (Campbell et al., 2005).
In the present study, JAK2 mutation was positive in 50% of patients with RARS-T and this was accompanied by rising platelet count. Similarly, Wang et al. (2006) found JAK2 exon 14 mutation among 50% of their RARS-T patients with a platelet count of >600 × 109/L. This clearly indicates that RARS-T has several features of MPN in addition to overproduction of platelets. There is some confusion as to the best cutoff platelet count level for a diagnosis of RARS-T when compared with RARS. The occurrence of JAK2 mutations and thrombocytosis in RARS is too frequent to be the result of chance only, and therefore, further studies are required to discover the mechanism of this association (Hellstrom-Lindberg and Cazzola, 2008).
In the present study, JAK2V617F mutation was not found among de novo AML, Ph+ CML, and MDS. Other studies reported the mutation in 2.7% of AML and 1.5% of MDS. Compared with the incidence of JAK2 mutation in PV, ET, and PMF, the incidence of JAK2 mutation in AML is very low (Kralovics et al., 2005; Lee et al., 2006).
During assay analysis for JAK2V617F mutational burden, differences were observed among patients: the majority was heterozygous for the mutation, whereas homozygosity was seen only among six patients (6.8%), four with PV and two with PMF. However, this has to be confirmed by more sensitive and accurate methodologies for quantification and separating homozygous from heterozygous JAK2V617F mutation, such as sequencing or quantitative real-time PCR (Merker et al., 2010).
The explanation of mutational burden variability is the differences in population size of mutant cells and variable ratio of heterozygous and homozygous cells for JAK2V617F. Allelic burden influences MPN-specific gene expression along with a number of clinical variables, such as secondary fibrosis, thrombosis, hematocrit, leukocytes, and platelet counts (Vannucchi et al., 2008). Presence of 9p unipaternal disomy resulting in gain of homozygosity along the short arm of chromosome 9 and gain of homozygosity for JAK2V617F is the main cause of high mutational burden in MPN (Campbell et al., 2006).
In the present study, white blood cell and platelet counts were found to be higher in patients with JAK2V617F mutation than those without mutation. Similar results were reported by Campbell et al. (2006).
The identification of JAK mutations has raised the exciting prospect of developing specific JAK2V617F inhibitors to treat PV patients. Given the remarkable clinical success of Imatinib therapy for the treatment of BCR-ABL-positive CML, FIP1L1-PDGFR-positive hypereosinophilic syndrome, and chronic myelomonocytic leukemia associated with platelet-derived growth factor beta (PDGFB) rearrangements and the development of second-generation BCR-ABL inhibitors for Imatinib refractory CML, the expectations that discovery of JAK2V617F will facilitate the development of targeted therapy for this mutation have been heightened. Recently, two tyrosine kinase inhibitors currently used clinically, Erlotinib and Cephalon-701, have been identified to inhibit JAK2V617F. Recent data suggest that interferon-α 2a and histone deacetylase inhibitors alone or in combination with JAK2 inhibitors may offer significant clinical benefit to patients with MPN (Liz et al., 2007; Hexner et al., 2008; Kiladjian et al., 2008).
Footnotes
Disclosure Statement
No competing financial interests exist.